Note: All my publications can also be found on Google Scholar
Authors: Giulio Isacchini, Takashi Okada, Clemens Schmid, Divya Ratan Popli, Benjamin Marco Peter, Stephan Schiffels and Oskar Hallatschek
Abstract:
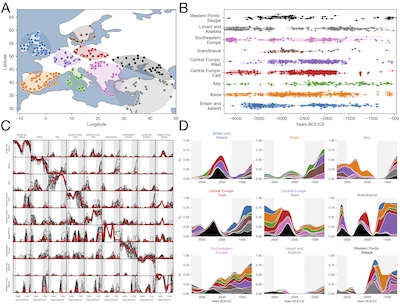
Major demographic events, such as population bottlenecks, founder effects, range expansions, and admixture events, have left lasting imprints on human genetic diversity. Ancient DNA (aDNA) sequencing now makes it increasingly possible to observe these signals across time, opening new avenues to address long-standing questions in human demographic history. Yet, deciphering this genetic archive of demographic history is challenging due to high levels of noise, and the complex ways in which demographic processes shape genetic variation. Here, leveraging the linear time evolution of the expectation of neutral allele frequencies, we develop a method to uncover systematic patterns of gene flow from metapopulation time-series data. We show that directional migration rates can be inferred via linear regression on time-dependent genetic dissimilarity between populations, quantified by an extended F 2 statistic evaluated between successive time points. Despite small sample sizes, the method reliably infers migration rates from simulated data by integrating information across multiple time slices. Applied to aDNA sampled from the last 6000 years, we recover signals of well-documented migrations and infer an ancient pan-European migration network. While complementing existing tools that estimate static ancestry proportions, our framework tracks how ancestry is dynamically redistributed through time.
Authors: Michał Golubiński, Mateusz Baca, Danijela Popović, Leo Speidel, Stephan Schiffels, Barbara Niezabitowska-Wiśniewska, Andrzej Kokowski and Martyna Molak
Abstract:
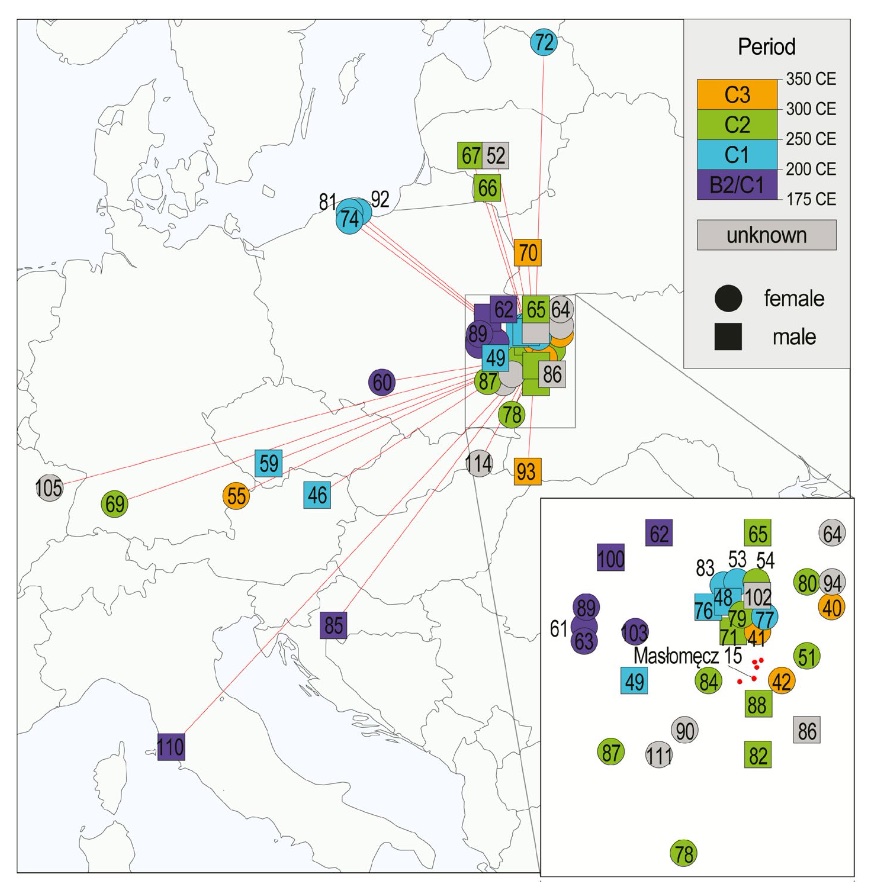
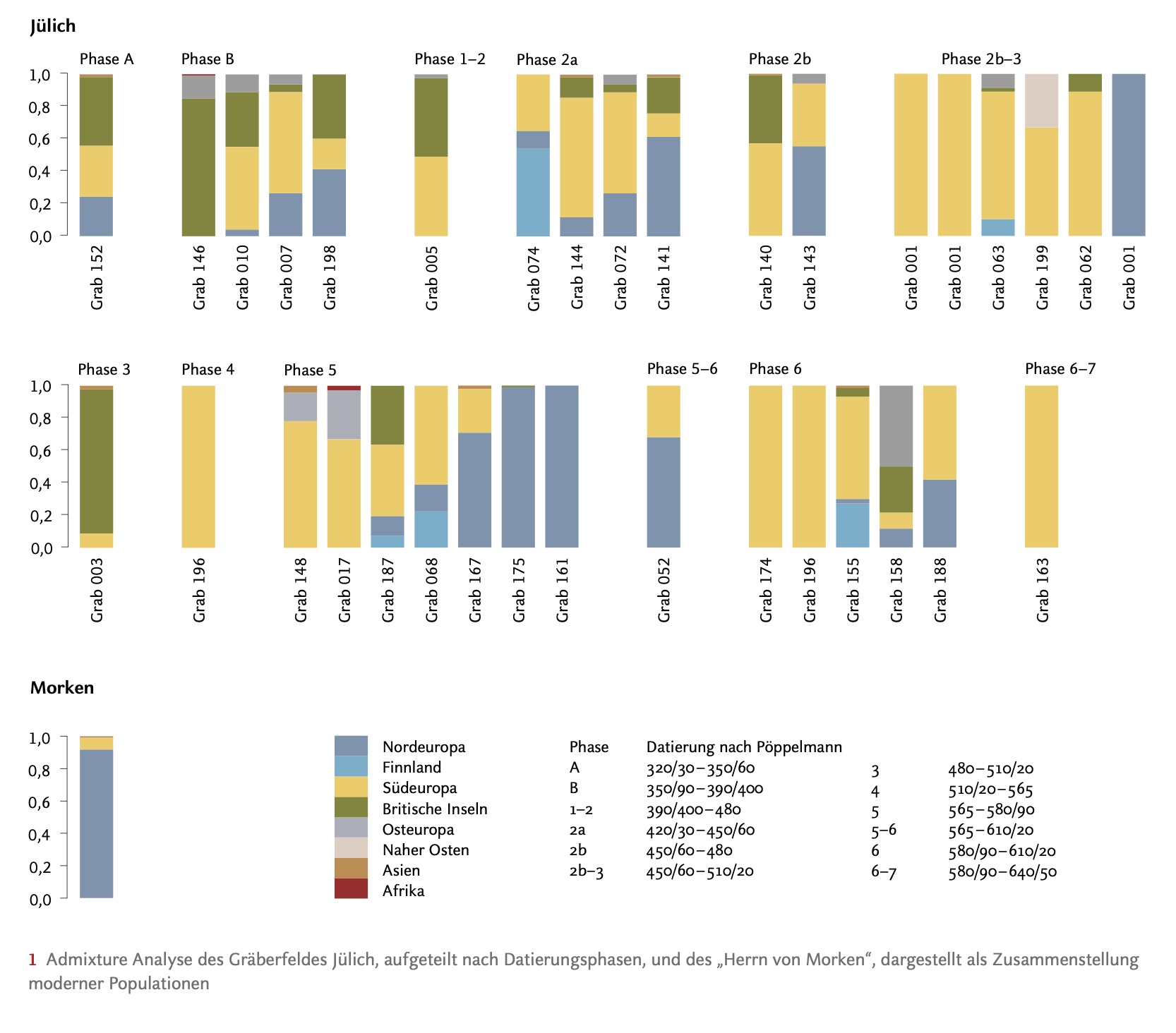
BACKGROUND: High mobility and extensive trade and military interactions are well recognized throughout the Late Iron Age Europe. The extremely rich archaeological record for the Masłomęcz group - a Goth-associated assemblage flourishing between 2nd and 4th century CE in what is now eastern Poland - has long been providing evidence for their wide cross-cultural contacts. However, the extent to which these were ephemeral or involved long-term immigration and interbreeding, remained unresolved. RESULTS: Here, by obtaining archaeogenomic data from 37 burials and reanalysing published data, we provide evidence that, while the Masłomęcz group was built mostly on Scandinavian-derived ancestry it extensively assimilated individuals from diverse directions and distances, including the Baltics, the Balkans and even further into the Mediterranean, creating a highly genetically heterogenous population. Additionally, we shed more light on the burial customs of this community by finding no close kin relations within multiperson burials. CONCLUSIONS: Our findings provide evidence for long-range mobility far outside the borders of the Roman Empire. The Masłomęcz group was a highly open community embracing external contacts and immigration, perhaps contradicting popular presumptions about the so-called barbarians.
Authors: Duncan Sayer, Joscha Gretzinger, John Hines, Michael McCormick, Keziah Warburton, E Sebo, Katharina Dulias, Maria Pala, Martin B Richards, C Edwards and S Schiffels
Abstract:
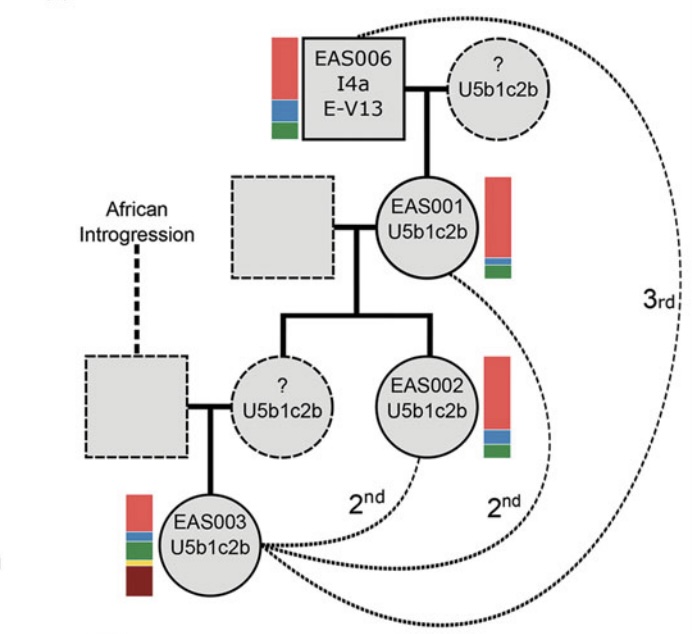
Archaeogenetics, the study of ancient DNA, can reveal powerful insights into kinship and the movement of individuals in (pre)history. Here, the authors report on the identification of two individuals with genetic profiles consistent with recent sub-Saharan African ancestry, both of whom were buried in early-medieval cemeteries in southern Britain. Focusing primarily on a sub-adult female from Updown in Kent, the authors explore the societal and cultural contexts in which these individuals lived and died, and the widening geographic links indicated by their presence, pointing back to the Byzantine reconquest of North Africa in AD 533--534.
Authors: M G B Foody, Katharina Dulias, P Justeau, Peter W Ditchfield, Lilian Ladle, Joscha Gretzinger, S Schiffels, David Reich, Robert A Kenyon, Duncan Sayer, Martin B Richards, Maria Pala and C Edwards
Abstract:
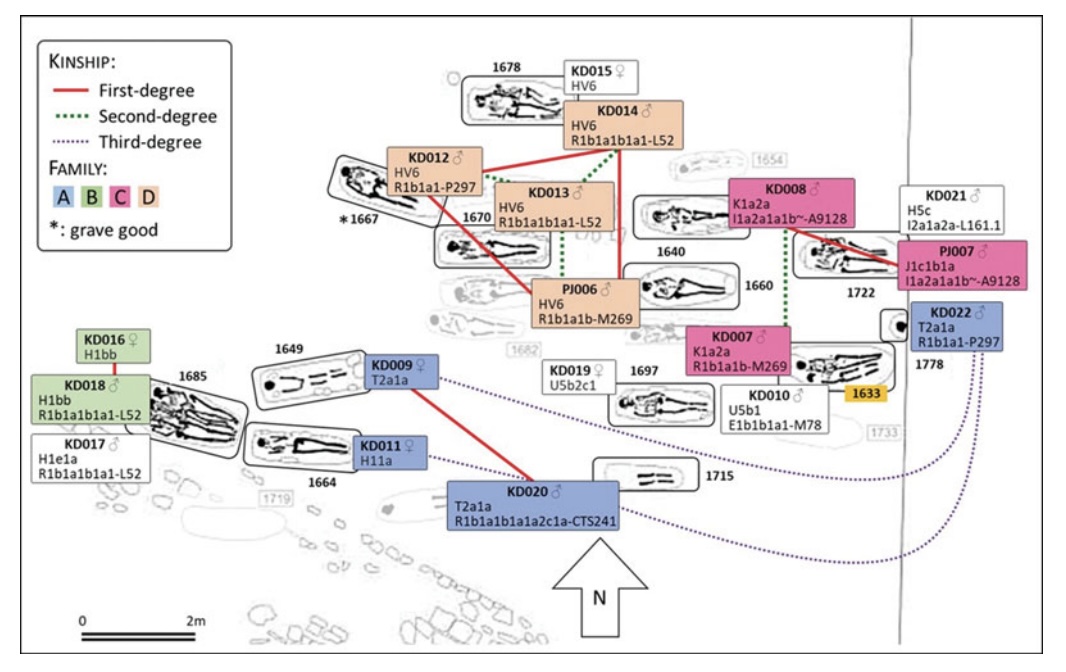
Kinship can be difficult to discern in the archaeological record, but the study of ancient DNA offers a useful window into one form of kinship: biological relatedness. Here, the authors explore possible kin connections at the post-Roman site of Worth Matravers in south-west England. They find that, while clusters of genetically related individuals are apparent, the inclusion of unrelated individuals in double or triple burials demonstrates an element of social kinship in burial location. Some individuals also carried genetic signatures of continental ancestry, with one young male revealing recent West African ancestry, highlighting the diverse heritage of early medieval Britain.
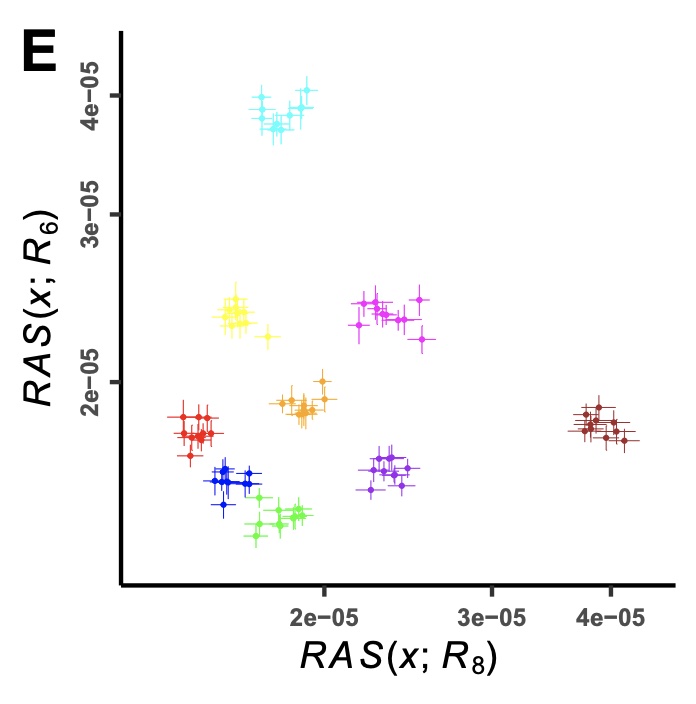
Authors: Lei Huang, Thiseas C Lamnidis and Stephan Schiffels
Abstract:
Various statistical methods have been developed to identify population structure from genetic data, including F-statistics, which measure the average correlation in allele frequency differences between two pairs of populations. However, the SNPs analyzed with F-statistics are often limited to those found as part of microarrays or, in the case of ancient DNA, to SNP capture panels, which are those within the common allele frequency band. Recent advances in sequencing technology increasingly allow generating whole-genome sequencing data, both ancient and modern, which not only enable querying nearly every base of the genome, but also contain numerous rare variants. Rare variants, with their more population-specific distribution, allow detection of population structure with much finer resolution than common variants - an opportunity that has so far been under-exploited. Here, we develop a new statistical method, RAS (Rare Allele Sharing), for summarizing rare allele frequency correlations, similar to F-statistics but with flexible ascertainment on allele frequencies. We test RAS on both published and simulated data and find that RAS has better resolution in distinguishing populations, with appropriate ascertainment. Leveraging this, we further develop the use of RAS to compute ancestry proportions with higher accuracy than existing methods, in cases of closely-related source populations. We implemented the new statistical methods as an R package and a command line tool. In summary, our method can provide new perspectives to identify and model population structure, allowing us to understand more subtle relationships among populations in the recent human past.

Authors: Felicitas Schmidt, Leif Hansen, Joscha Gretzinger, Angela Mötsch, Stephan Schiffels and Dirk Krausse
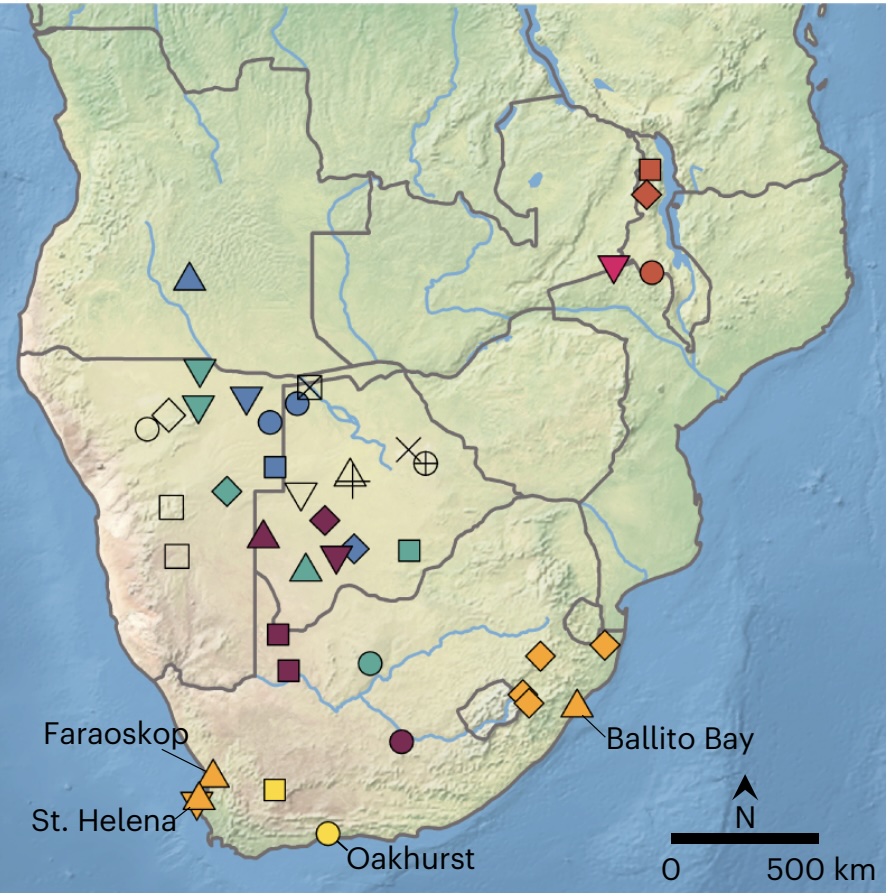
Authors: Joscha Gretzinger, Victoria E Gibbon, Sandra E Penske, Judith C Sealy, Adam B Rohrlach, Domingo C Salazar-Garcia, Johannes Krause and Stephan Schiffels
Abstract:
Southern Africa has one of the longest records of fossil hominins and harbours the largest human genetic diversity in the world. Yet, despite its relevance for human origins and spread around the globe, the formation and processes of its gene pool in the past are still largely unknown. Here, we present a time transect of genome-wide sequences from nine individuals recovered from a single site in South Africa, Oakhurst Rockshelter. Spanning the whole Holocene, the ancient DNA of these individuals allows us to reconstruct the demographic trajectories of the indigenous San population and their ancestors during the last 10,000 years. We show that, in contrast to most regions around the world, the population history of southernmost Africa was not characterized by several waves of migration, replacement and admixture but by long-lasting genetic continuity from the early Holocene to the end of the Later Stone Age. Although the advent of pastoralism and farming substantially transformed the gene pool in most parts of southern Africa after 1,300 bp, we demonstrate using allele-frequency and identity-by-descent segment-based methods that the \textdaggerdbl{}Khomani San and Karretjiemense from South Africa still show direct signs of relatedness to the Oakhurst hunter-gatherers, a pattern obscured by recent, extensive non-Southern African admixture. Yet, some southern San in South Africa still preserve this ancient, Pleistocene-derived genetic signature, extending the period of genetic continuity until today.
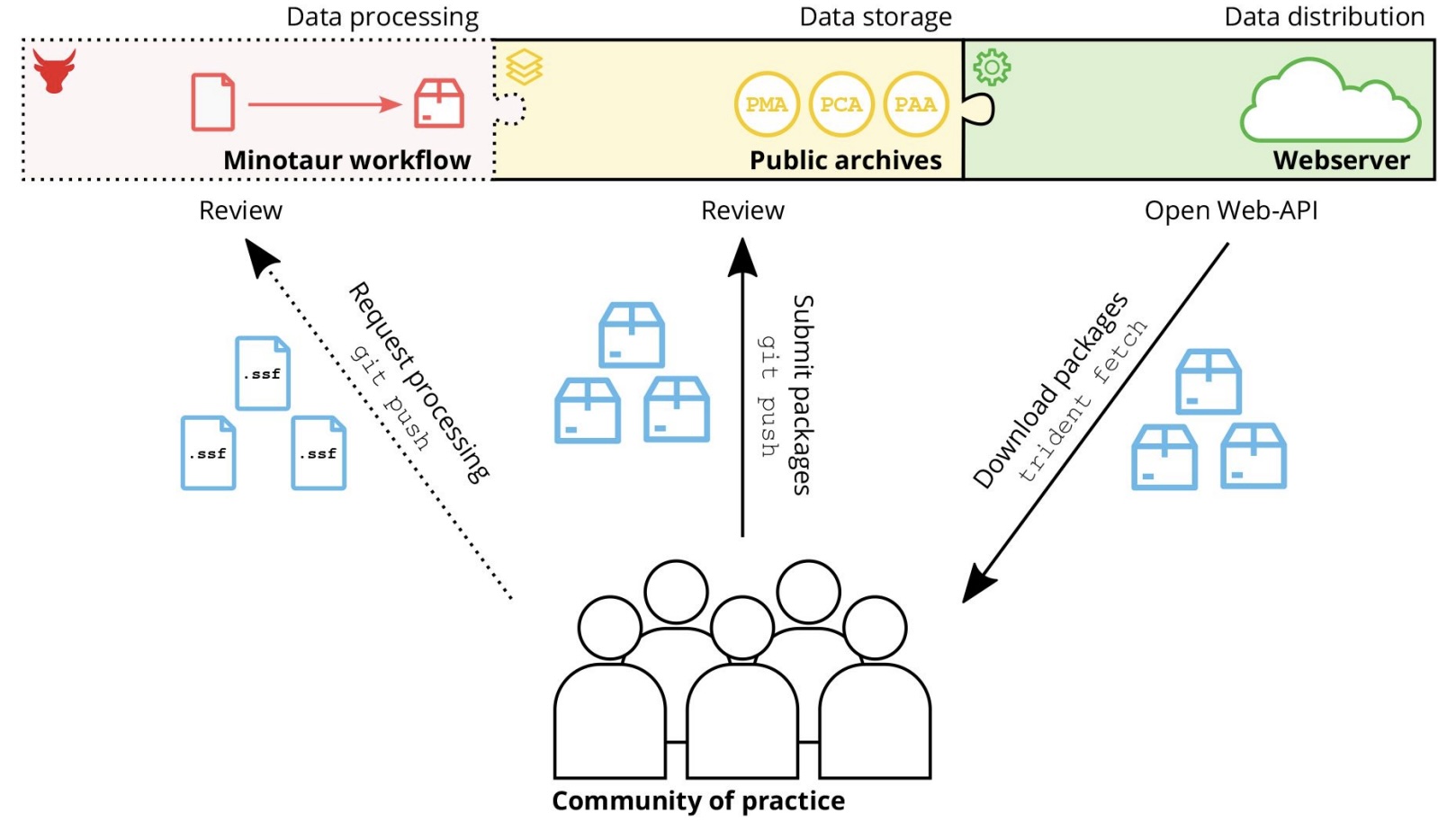
Authors: Clemens Schmid, Ayshin Ghalichi, Thiseas C Lamnidis, Dhananjaya B A Mudiyanselage, Wolfgang Haak and Stephan Schiffels
Abstract:
The study of ancient human genomes, archaeo- or palaeogenetics, has accelerated in the last ten years, with now thousands of new ancient genomes being released each year. Operating at the interface of genetics, anthro-pology and archaeology, this data includes features from all three fields, including rich meta- and context-data, for example regarding spatiotemporal provenience. While archives and standards for genetic sequencing data al-ready exist, no such infrastructure exists for combined genetic and meta-data that could ensure FAIR principles across the field. Here, we present Poseidon, a framework for open and FAIR data handling in archaeogenetics, including a specified package format, software tools, and public, community-maintained online archives. Poseidon emphasises human- and machine-readable data storage, the development of convenient and interoperable command line software, and a high degree of source granularity to elevate the original data publication to the main unit of long-term curation.
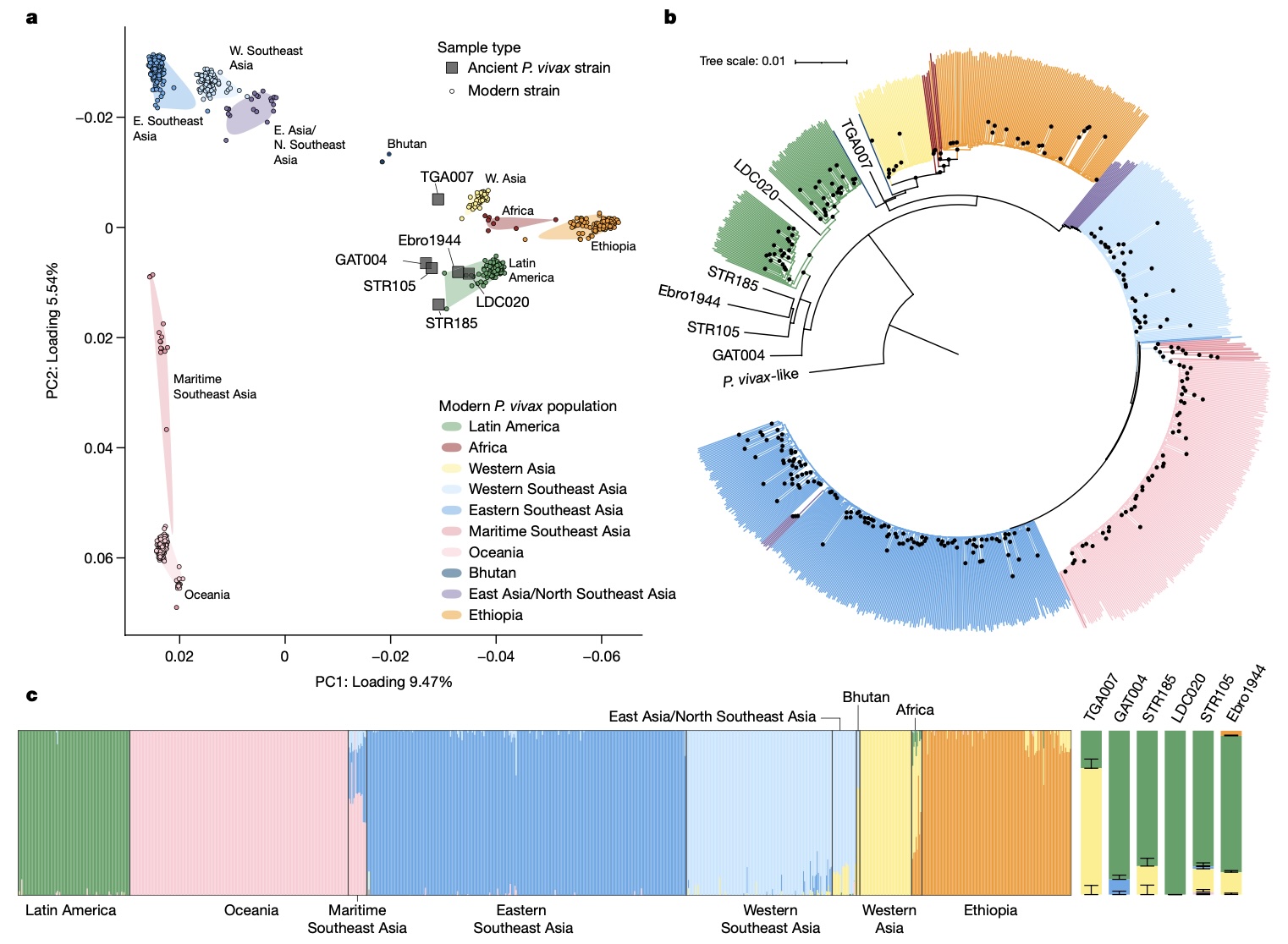
Authors: Megan Michel, Eirini Skourtanioti, Federica Pierini, Evelyn K Guevara, Angela Mötsch, Arthur Kocher, Rodrigo Barquera, Raffaela A Bianco, Selina Carlhoff, Lorenza Coppola Bove, Suzanne Freilich, Karen Giffin, Taylor Hermes, Alina Hiß, Florian Knolle, Elizabeth A Nelson, Gunnar U Neumann, Luka Papac, Sandra Penske, Adam B Rohrlach, Nada Salem, Lena Semerau, Vanessa Villalba-Mouco, Isabelle Abadie, Mark Aldenderfer, Jessica F Beckett, Matthew Brown, Franco G R Campus, Tsang Chenghwa, María Cruz Berrocal, Ladislav Damašek, Kellie Sara Duffett Carlson, Raphaël Durand, Michal Ernée, Cristinel Fântăneanu, Hannah Frenzel, Gabriel García Atiénzar, Sonia Guillén, Ellen Hsieh, Maciej Karwowski, David Kelvin, Nikki Kelvin, Alexander Khokhlov, Rebecca L Kinaston, Arkadii Korolev, Kim-Louise Krettek, Mario Küßner, Luca Lai, Cory Look, Kerttu Majander, Kirsten Mandl, Vittorio Mazzarello, Michael McCormick, Patxuka de Miguel Ibáñez, Reg Murphy, Rita E Németh, Kerkko Nordqvist, Friederike Novotny, Martin Obenaus, Lauro Olmo-Enciso, Päivi Onkamo, Jörg Orschiedt, Valerii Patrushev, Sanni Peltola, Alejandro Romero, Salvatore Rubino, Antti Sajantila, Domingo C Salazar-García, Elena Serrano, Shapulat Shaydullaev, Emanuela Sias, Mario Šlaus, Ladislav Stančo, Treena Swanston, Maria Teschler-Nicola, Frederique Valentin, Katrien Van de Vijver, Tamara L Varney, Alfonso Vigil-Escalera Guirado, Christopher K Waters, Estella Weiss-Krejci, Eduard Winter, Thiseas C Lamnidis, Kay Prüfer, Kathrin Nägele, Maria Spyrou, Stephan Schiffels, Philipp W Stockhammer, Wolfgang Haak, Cosimo Posth, Christina Warinner, Kirsten I Bos, Alexander Herbig and Johannes Krause
Abstract:
Malaria-causing protozoa of the genus Plasmodium have exerted one of the strongest selective pressures on the human genome, and resistance alleles provide biomolecular footprints that outline the historical reach of these species1. Nevertheless, debate persists over when and how malaria parasites emerged as human pathogens and spread around the globe1,2. To address these questions, we generated high-coverage ancient mitochondrial and nuclear genome-wide data from P. falciparum, P. vivax and P. malariae from 16 countries spanning around 5,500 years of human history. We identified P. vivax and P. falciparum across geographically disparate regions of Eurasia from as early as the fourth and first millennia BCE, respectively; for P. vivax, this evidence pre-dates textual references by several millennia3. Genomic analysis supports distinct disease histories for P. falciparum and P. vivax in the Americas: similarities between now-eliminated European and peri-contact South American strains indicate that European colonizers were the source of American P. vivax, whereas the trans-Atlantic slave trade probably introduced P. falciparum into the Americas. Our data underscore the role of cross-cultural contacts in the dissemination of malaria, laying the biomolecular foundation for future palaeo-epidemiological research into the impact of Plasmodium parasites on human history. Finally, our unexpected discovery of P. falciparum in the high-altitude Himalayas provides a rare case study in which individual mobility can be inferred from infection status, adding to our knowledge of cross-cultural connectivity in the region nearly three millennia ago.
Authors: Ainash Childebayeva, Fabian Fricke, A Rohrlach, Lei Huang, S Schiffels, O Vesakoski, K Mannermaa, Lena Semerau, Franziska Aron, K Solodovnikov, M Rykun, V Moiseyev, V Khartanovich, Igor Kovtun, Johannes Krause, Sergey Kuzminykh and W Haak
Abstract:
The Eurasian Bronze Age (BA) has been described as a period of substantial human migrations, the emergence of pastoralism, horse domestication, and development of metallurgy. This study focuses on two north Eurasian sites sharing Siberian genetic ancestry. One of the sites, Rostovka, is associated with the Seima-Turbino (ST) phenomenon (~2200-1900 BCE) that is characterized by elaborate metallurgical objects found throughout Northern Eurasia. The genetic profiles of Rostovka individuals vary widely along the forest-tundra Siberian genetic cline represented by many modern Uralic-speaking populations, and the genetic heterogeneity observed is consistent with the current understanding of the ST being a transcultural phenomenon. Individuals from the second site, Bolshoy Oleni Ostrov in Kola, in comparison form a tighter cluster on the Siberian ancestry cline. We further explore this Siberian ancestry profile and assess the role of the ST phenomenon and other contemporaneous BA cultures in the spread of Uralic languages and Siberian ancestry.
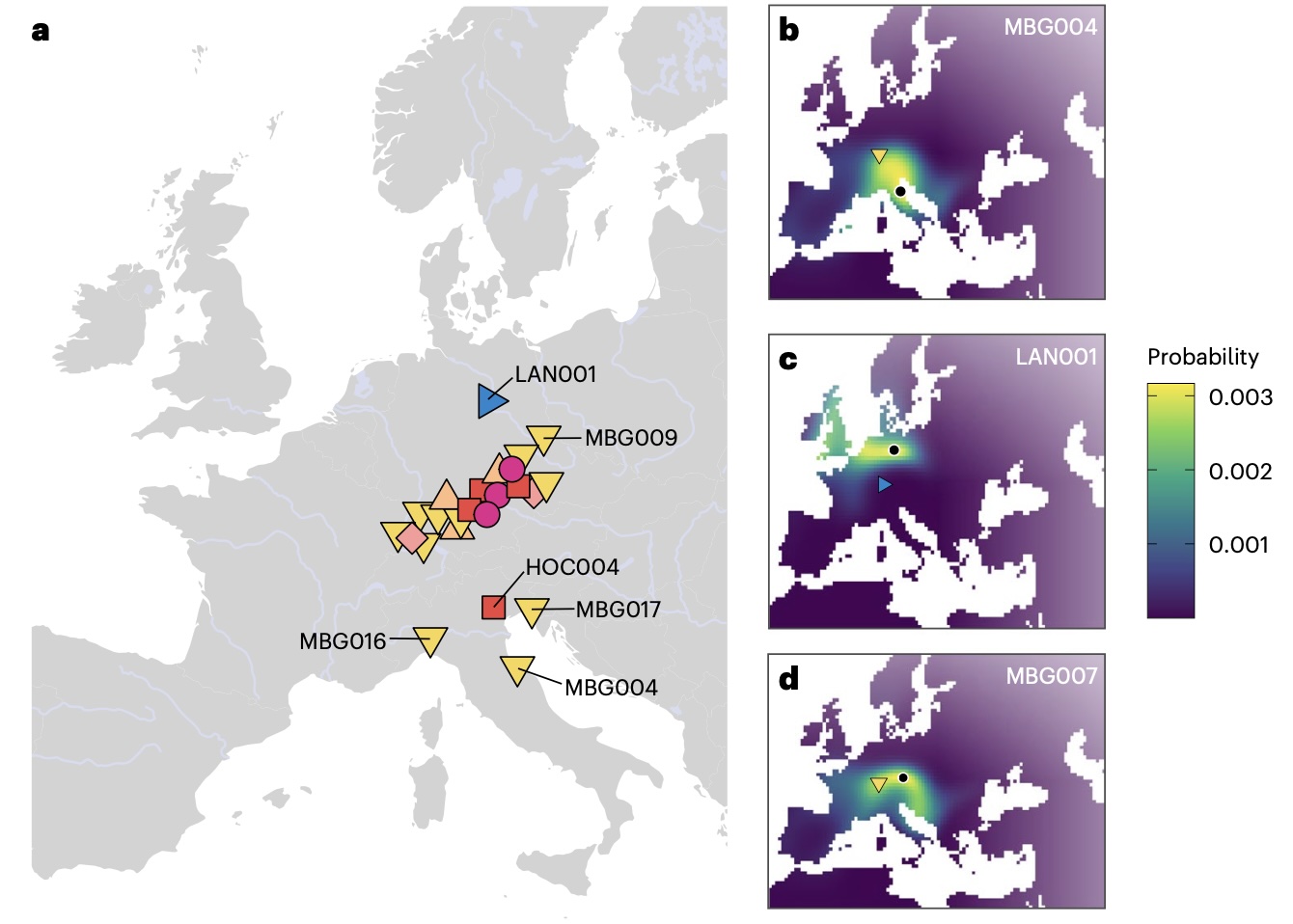
Authors: Joscha Gretzinger, Felicitas Schmitt, Angela Mötsch, Selina Carlhoff, Thiseas Christos Lamnidis, Yilei Huang, Harald Ringbauer, Corina Knipper, Michael Francken, Franziska Mandt, Leif Hansen, Cäcilia Freund, Hannes Rathmann, Katerina Harvati, Günther Wieland, Lena Granehäll, Frank Maixner, Albert Zink, Wolfram Schier, Dirk Krausse, Johannes Krause and Stephan Schiffels
Abstract:
The early Iron Age (800 to 450 BCE) in France, Germany and Switzerland, known as the `West-Hallstattkreis', stands out as featuring the earliest evidence for supra-regional organization north of the Alps. Often referred to as `early Celtic', suggesting tentative connections to later cultural phenomena, its societal and population structure remain enigmatic. Here we present genomic and isotope data from 31 individuals from this context in southern Germany, dating between 616 and 200 BCE. We identify multiple biologically related groups spanning three elite burials as far as 100 km apart, supported by trans-regional individual mobility inferred from isotope data. These include a close biological relationship between two of the richest burial mounds of the Hallstatt culture. Bayesian modelling points to an avuncular relationship between the two individuals, which may suggest a practice of matrilineal dynastic succession in early Celtic elites. We show that their ancestry is shared on a broad geographic scale from Iberia throughout Central-Eastern Europe, undergoing a decline after the late Iron Age (450 BCE to ~50 CE). Gretzinger et al. examine genetic evidence from 31 Iron Age individuals in southern Germany and find that this early Celtic society probably had a dynastic system of matrilineal inheritance, with a network of well-connected elites covering a broad territory.
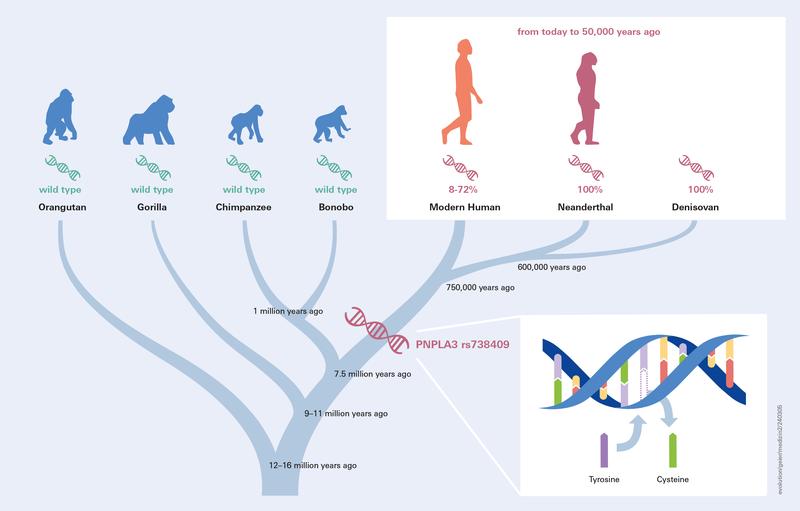
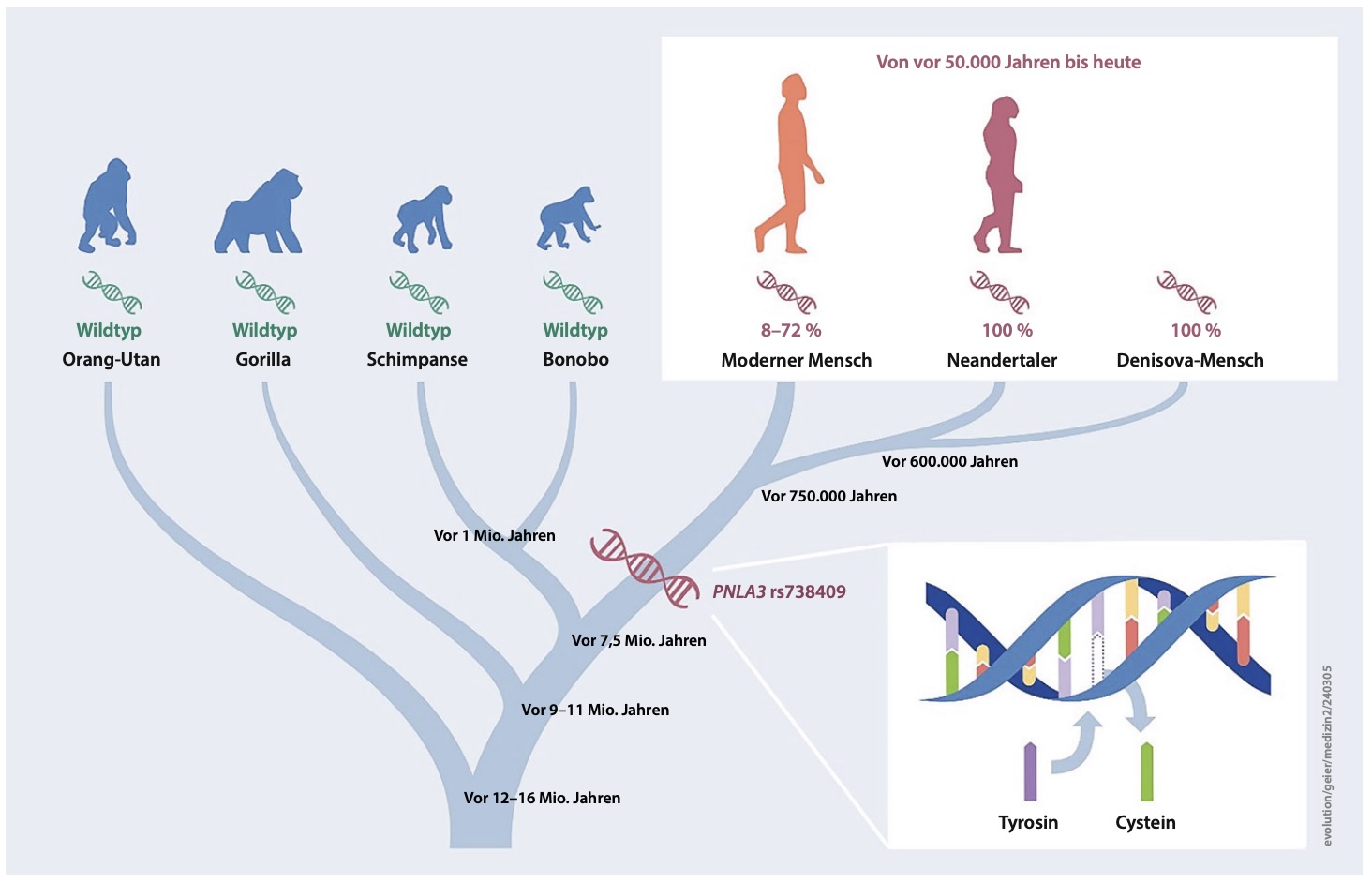
Authors: Andreas Geier, Jonas Trost, Ke Wang, Clemens Schmid, Marcin Krawczyk and Stephan Schiffels
Abstract:
OBJECTIVE: Fat deposition is modulated by environmental factors and genetic predisposition. Genome-wide association studies identified PNPLA3 p.I148M (rs738409) as a common variant that increases risk of developing liver steatosis. When and how this variant evolved in humans has not been studied to date. DESIGN: Here we analyse ancient DNA to track the history of this allele throughout human history. In total, 6444 published ancient (modern humans, Neanderthal, Denisovan) and 3943 published present day genomes were used for analysis after extracting genotype calls for PNPLA3 p.I148M. To quantify changes through time, logistic and, by grouping individuals according to geography and age, linear regression analyses were performed. RESULTS: We find that archaic human individuals (Neanderthal, Denisovan) exclusively carried a fixed PNPLA3 risk allele, whereas allele frequencies in modern human populations range from very low in Africa to >50\% in Mesoamerica. Over the last 15 000 years, distributions of ancestral and derived alleles roughly match the present day distribution. Logistic regression analyses did not yield signals of natural selection during the last 10 000 years. CONCLUSION: Archaic human individuals exclusively carried a fixed PNPLA3 allele associated with fatty liver, whereas allele frequencies in modern human populations are variable even in the oldest samples. Our observation might underscore the advantage of fat storage in cold climate and particularly for Neanderthal under ice age conditions. The absent signals of natural selection during modern human history does not support the thrifty gene hypothesis in case of PNPLA3 p.I148M.
Authors: Adam Benjamin Rohrlach, Maïté Rivollat, Patxuka de-Miguel-Ibáñez, Ulla Moilanen, Anne-Mari Liira, João C Teixeira, Xavier Roca-Rada, Javier Armendáriz-Martija, Kamen Boyadzhiev, Yavor Boyadzhiev, Bastien Llamas, Anthi Tiliakou, Angela Mötsch, Jonathan Tuke, Eleni-Anna Prevedorou, Naya Polychronakou-Sgouritsa, Jane Buikstra, Päivi Onkamo, Philipp W Stockhammer, Henrike O Heyne, Johannes R Lemke, Roberto Risch, Stephan Schiffels, Johannes Krause, Wolfgang Haak and Kay Prüfer
Abstract:
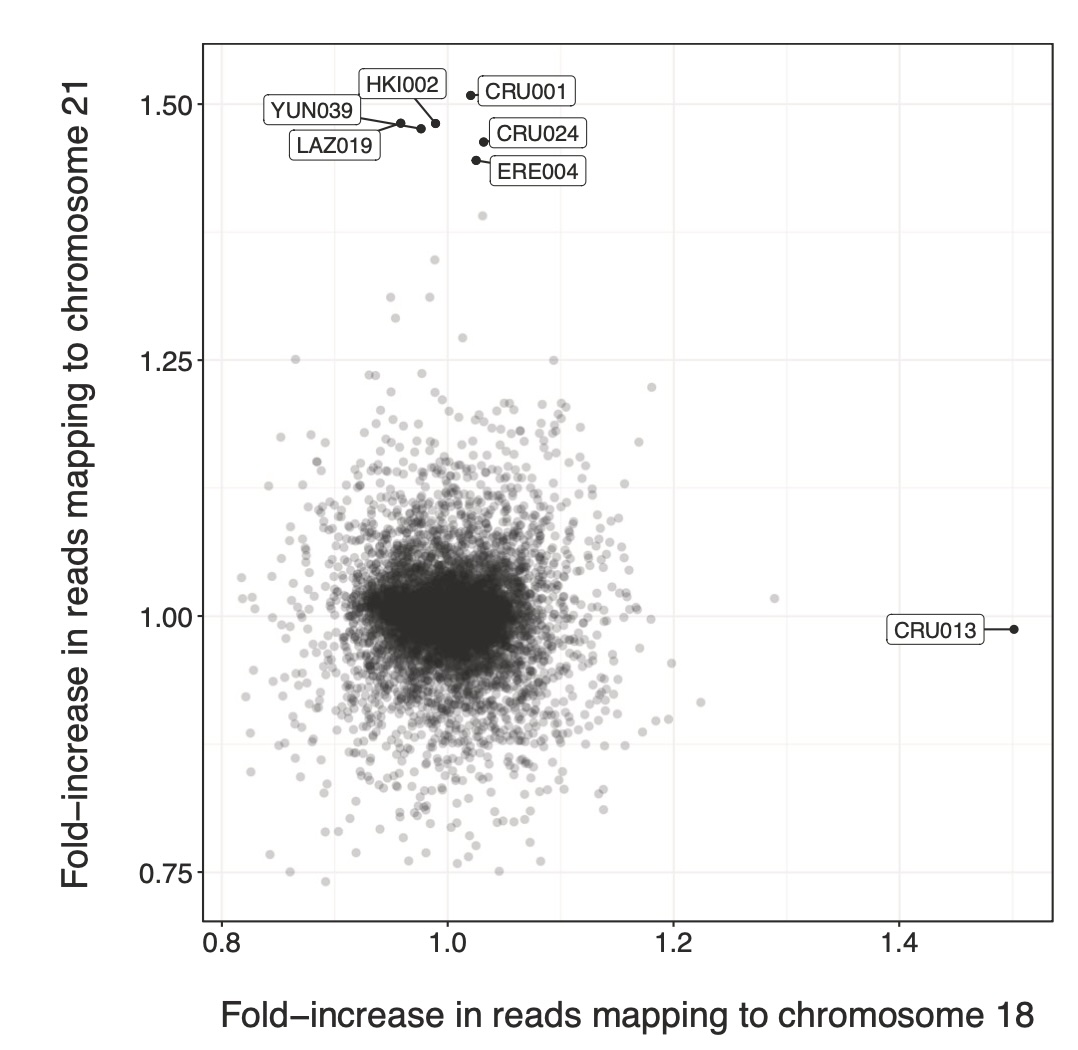
Aneuploidies, and in particular, trisomies represent the most common genetic aberrations observed in human genetics today. To explore the presence of trisomies in historic and prehistoric populations we screen nearly 10,000 ancient human individuals for the presence of three copies of any of the target autosomes. We find clear genetic evidence for six cases of trisomy 21 (Down syndrome) and one case of trisomy 18 (Edwards syndrome), and all cases are present in infant or perinatal burials. We perform comparative osteological examinations of the skeletal remains and find overlapping skeletal markers, many of which are consistent with these syndromes. Interestingly, three cases of trisomy 21, and the case of trisomy 18 were detected in two contemporaneous sites in early Iron Age Spain (800-400 BCE), potentially suggesting a higher frequency of burials of trisomy carriers in those societies. Notably, the care with which the burials were conducted, and the items found with these individuals indicate that ancient societies likely acknowledged these individuals with trisomy 18 and 21 as members of their communities, from the perspective of burial practice.

Authors: Luka Papac, Patxuka de Miguel Ibáñez, Adam B Rohrlach, Javier Armendáriz, Marcello Peres, Thiseas C Lamnidis, Angela Mötsch, Stephan Schiffels and Roberto Risch
Abstract:
The transition from the Late Bronze to the Iron Age on the Iberian Peninsula saw a shift in mortuary customs from mainly inhumation to cremation of the deceased. The poor preservation characteristic of cremated skeletal remains has hindered molecular analyses (isotope analyses, ancient DNA) of the Iberian Final Bronze and Iron Age communities of Ibe- ria. Incidentally, a limited number of young children, often newborns, were exempt from the predominant cremation ritual, in favour of intramural inhumations inside buildings at certain settlements. The discourse surrounding the mean- ing and interpretation of this particular burial rite has devel- oped over a long time in Iberian archaeology but has always been hampered by the limited anthropological, archaeologi- cal, and molecular data from these intramural inhumations. Here, we study the genomes of 37 intramurally buried chil- dren found in three Early Iron Age settlements, dated between c. 800--450 BC. Population genetic analyses on the newly reported individuals extend our understanding of ancient Iberia by revealing previously unsampled genetic diversity as well as showing a lesser influence of Mediterranean ancestry than on previously published Iron Age individuals from north- ern Spain. We also provide insights into the sex and biological relatedness of the children, and in so doing, elucidate differ- ent aspects of the intramural burial ritual and building use in settlements. More broadly, the genetic data from these indi- viduals fill an important gap in the archaeogenetic record of northern Spain and offer a unique opportunity to study the genetic makeup and population changes from the Bronze Age to Antiquity.
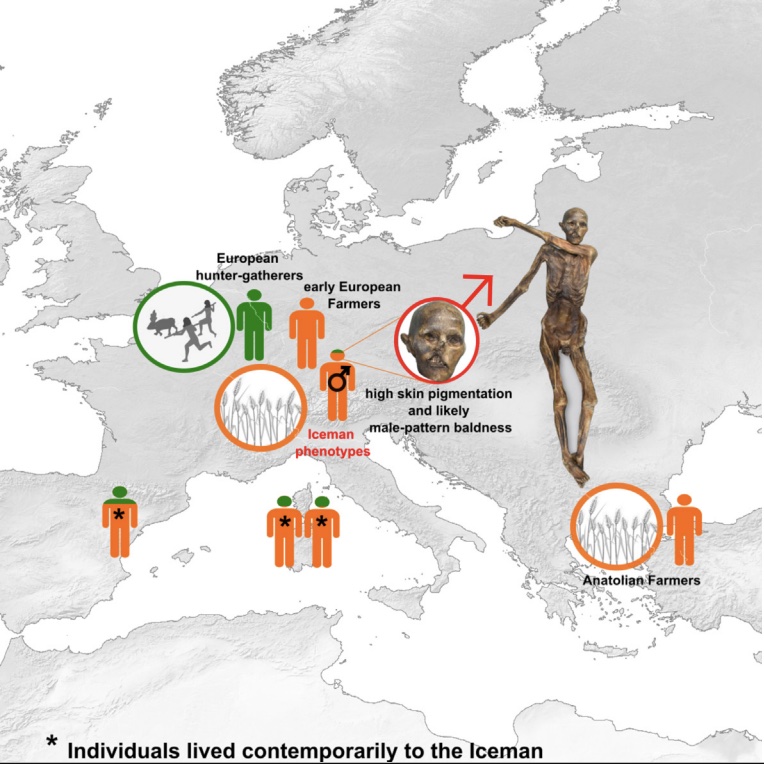
Authors: Ke Wang, Kay Prüfer, Ben Krause-Kyora, Ainash Childebayeva, Verena J Schuenemann, Valentina Coia, Frank Maixner, Albert Zink, Stephan Schiffels and Johannes Krause
Abstract:
The Tyrolean Iceman is known as one of the oldest human glacier mummies, directly dated to 3350–3120 calibrated BCE. A previously published low-coverage genome provided novel insights into European prehistory, despite high present-day DNA contamination. Here, we generate a high-coverage genome with low contamination (15.3×) to gain further insights into the genetic history and phenotype of this individual. Contrary to previous studies, we found no detectable Steppe-related ancestry in the Iceman. Instead, he retained the highest Anatolian-farmer-related ancestry among contemporaneous European populations, indicating a rather isolated Alpine population with limited gene flow from hunter-gatherer-ancestry-related populations. Phenotypic analysis revealed that the Iceman likely had darker skin than present-day Europeans and carried risk alleles associated with male-pattern baldness, type 2 diabetes, and obesity-related metabolic syndrome. These results corroborate phenotypic observations of the preserved mummified body, such as high pigmentation of his skin and the absence of hair on his head.

Authors: Victoria E Gibbon, Loretta Feris, Joscha Gretzinger, Kathryn Smith, Simon Hall, Nigel Penn, Tinashe E M Mutsvangwa, Michaela Heale, Devin A Finaughty, Yvonne W Karanja, Jan Esterhuyse, Daniël Kotze, Nina Barnes, Geney Gunston, Je'nine May, Johannes Krause, Caroline M Wilkinson, Stephan Schiffels, Doreen Februarie, Sianne Alves and Judith C Sealy
Abstract:
We describe a process of restitution of nine unethically acquired human skeletons to their families, together with attempts at redress. Between 1925-1927 C.E., the skeletonised remains of nine San or Khoekhoe people, eight of them known-in-life, were removed from their graves on the farm Kruisrivier, near Sutherland in the Northern Cape Province of South Africa. They were donated to the Anatomy Department at the University of Cape Town. This was done without the knowledge or permission of their families. The donor was a medical student who removed the remains from the labourers' cemetery on his family farm. Nearly 100 years later, the remains are being returned to their community, accompanied by a range of community-driven interdisciplinary historical, archaeological and analytical (osteobiographic, craniofacial, ancient DNA, stable isotope) studies to document, as far as possible, their lives and deaths. The restitution process began by contacting families living in the same area with the same surnames as the deceased. The restitution and redress process prioritises the descendant families' memories, wishes and desire to understand the situation, and learn more about their ancestors. The descendant families have described the process as helping them to reconnect with their ancestors. A richer appreciation of their ancestors' lives, gained in part from scientific analyses, culminating with reburial, is hoped to aid the descendant families and wider community in [re-]connecting with their heritage and culture, and contribute to restorative justice, reconciliation and healing while confronting a traumatic historical moment. While these nine individuals were exhumed as specimens, they will be reburied as people.
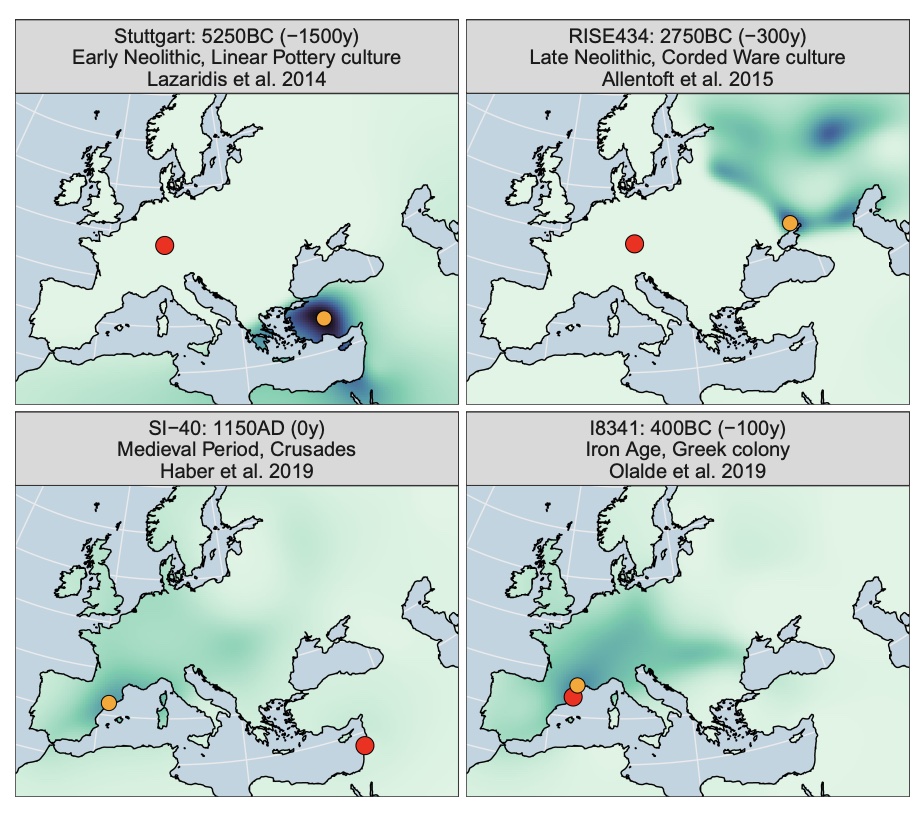
Authors: Clemens Schmid and Stephan Schiffels
Abstract:
The recent increase in openly available ancient human DNA samples allows for large-scale meta-analysis applications. Trans-generational past human mobility is one of the key aspects that ancient genomics can contribute to since changes in genetic ancestry-unlike cultural changes seen in the archaeological record-necessarily reflect movements of people. Here, we present an algorithm for spatiotemporal mapping of genetic profiles, which allow for direct estimates of past human mobility from large ancient genomic datasets. The key idea of the method is to derive a spatial probability surface of genetic similarity for each individual in its respective past. This is achieved by first creating an interpolated ancestry field through space and time based on multivariate statistics and Gaussian process regression and then using this field to map the ancient individuals into space according to their genetic profile. We apply this algorithm to a dataset of 3138 aDNA samples with genome-wide data from Western Eurasia in the last 10,000 y. Finally, we condense this sample-wise record with a simple summary statistic into a diachronic measure of mobility for subregions in Western, Central, and Southern Europe. For regions and periods with sufficient data coverage, our similarity surfaces and mobility estimates show general concordance with previous results and provide a meta-perspective of genetic changes and human mobility.
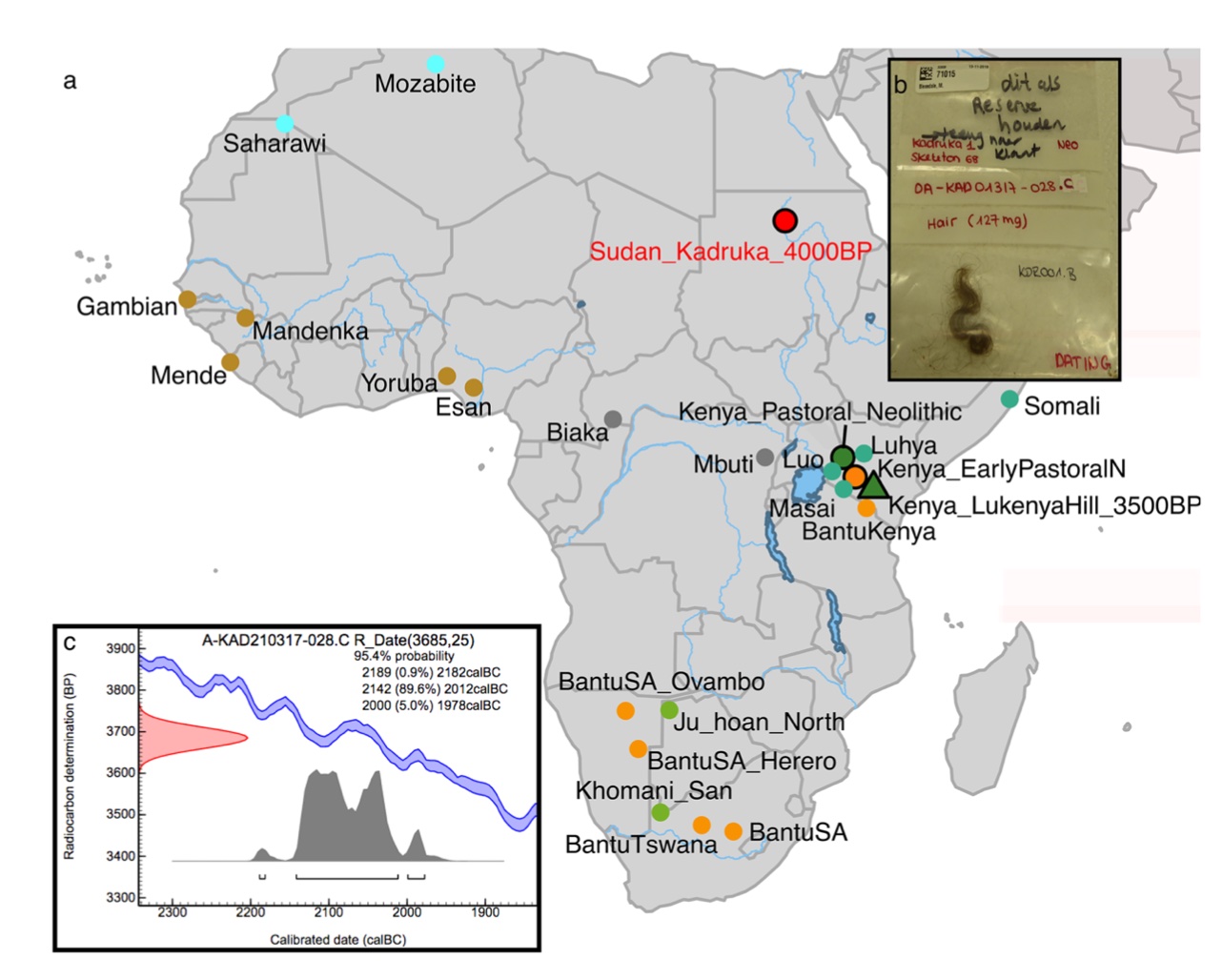
Authors: Ke Wang, Madeleine Bleasdale, Charles Le Moyne, Cacilia Freund, Johannes Krause, Nicole Boivin and Stephan Schiffels
Abstract:
Petrous bones and teeth are the skeletal elements most often targeted by researchers for ancient DNA (aDNA) extraction, and the sources of the majority of previously published ancient African genomes. However, the high temperature environments that characterise much of Africa often lead to poor preservation of skeletal remains. Here, we successfully reconstruct and analyse genome-wide data from the naturally mummified hair of a 4000-year-old individual from Sudan in northeastern Africa, after failed attempts at DNA extraction from teeth, petrous, and cranium of this and other individuals from the Kadruka cemeteries. We find that hair DNA extracted with an established single-stranded library protocol is unusually enriched in ultra-short DNA molecules and exhibits substantial interior molecular damage. The aDNA was nonetheless amenable to genetic analyses, which revealed that the genome is genetically indistinguishable from that of early Neolithic eastern African pastoralists located 2500 kms away. Our findings are consistent with established models for the southward dispersal of Middle Nile Valley pastoral populations to the Rift Valley of eastern Africa, and provide a possible genetic source population for this dispersal. Our study highlights the value of mummified hair as an alternate source of aDNA from regions with poor bone preservation.
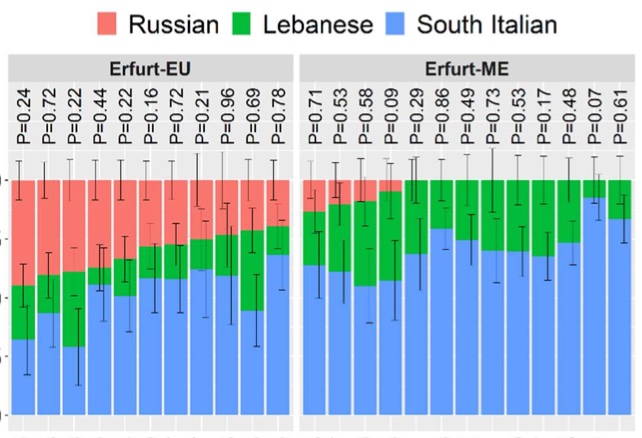
Authors: Shamam Waldman, Daniel Backenroth, Éadaoin Harney, Stefan Flohr, Nadia C Neff, Gina M Buckley, Hila Fridman, Ali Akbari, Nadin Rohland, Swapan Mallick, Iñigo Olalde, Leo Cooper, Ariel Lomes, Joshua Lipson, Jorge Cano Nistal, Jin Yu, Nir Barzilai, Inga Peter, Gil Atzmon, Harry Ostrer, Todd Lencz, Yosef E Maruvka, Maike Lämmerhirt, Alexander Beider, Leonard V Rutgers, Virginie Renson, Keith M Prufer, Stephan Schiffels, Harald Ringbauer, Karin Sczech, Shai Carmi and David Reich
Abstract:
We report genome-wide data from 33 Ashkenazi Jews (AJ), dated to the 14th century, obtained following a salvage excavation at the medieval Jewish cemetery of Erfurt, Germany. The Erfurt individuals are genetically similar to modern AJ, but they show more variability in Eastern European-related ancestry than modern AJ. A third of the Erfurt individuals carried a mitochondrial lineage common in modern AJ and eight carried pathogenic variants known to affect AJ today. These observations, together with high levels of runs of homozygosity, suggest that the Erfurt community had already experienced the major reduction in size that affected modern AJ. The Erfurt bottleneck was more severe, implying substructure in medieval AJ. Overall, our results suggest that the AJ founder event and the acquisition of the main sources of ancestry pre-dated the 14th century and highlight late medieval genetic heterogeneity no longer present in modern AJ.

Authors: Joscha Gretzinger and Stephan Schiffels
Abstract:
Ancient human DNA is revolutionising our ability to detect human migration that occurred hundreds of years ago. Joscha Gretzinger and Stephan Schiffels explain how these advances have transformed our understanding of the past
Authors: Joscha Gretzinger and Stephan Schiffels
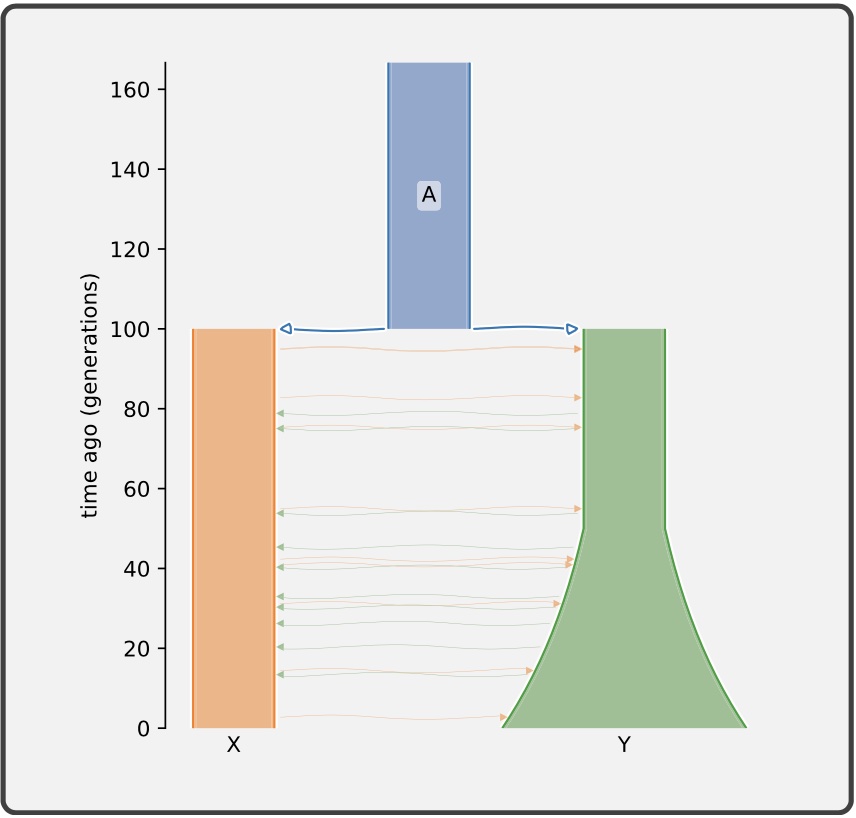
Authors: Graham Gower, Aaron P Ragsdale, Gertjan Bisschop, Ryan N Gutenkunst, Matthew Hartfield, Ekaterina Noskova, Stephan Schiffels, Travis J Struck, Jerome Kelleher and Kevin R Thornton
Abstract:
Understanding the demographic history of populations is a key goal in population genetics, and with improving methods and data, ever more complex models are being proposed and tested. Demographic models of current interest typically consist of a set of discrete populations, their sizes and growth rates, and continuous and pulse migrations between those populations over a number of epochs, which can require dozens of parameters to fully describe. There is currently no standard format to define such models, significantly hampering progress in the field. In particular, the important task of translating the model descriptions in published work into input suitable for population genetic simulators is labor intensive and error prone. We propose the Demes data model and file format, built on widely used technologies, to alleviate these issues. Demes provide a well-defined and unambiguous model of populations and their properties that is straightforward to implement in software, and a text file format that is designed for simplicity and clarity. We provide thoroughly tested implementations of Demes parsers in multiple languages including Python and C, and showcase initial support in several simulators and inference methods. An introduction to the file format and a detailed specification are available at https://popsim-consortium.github.io/demes-spec-docs/.
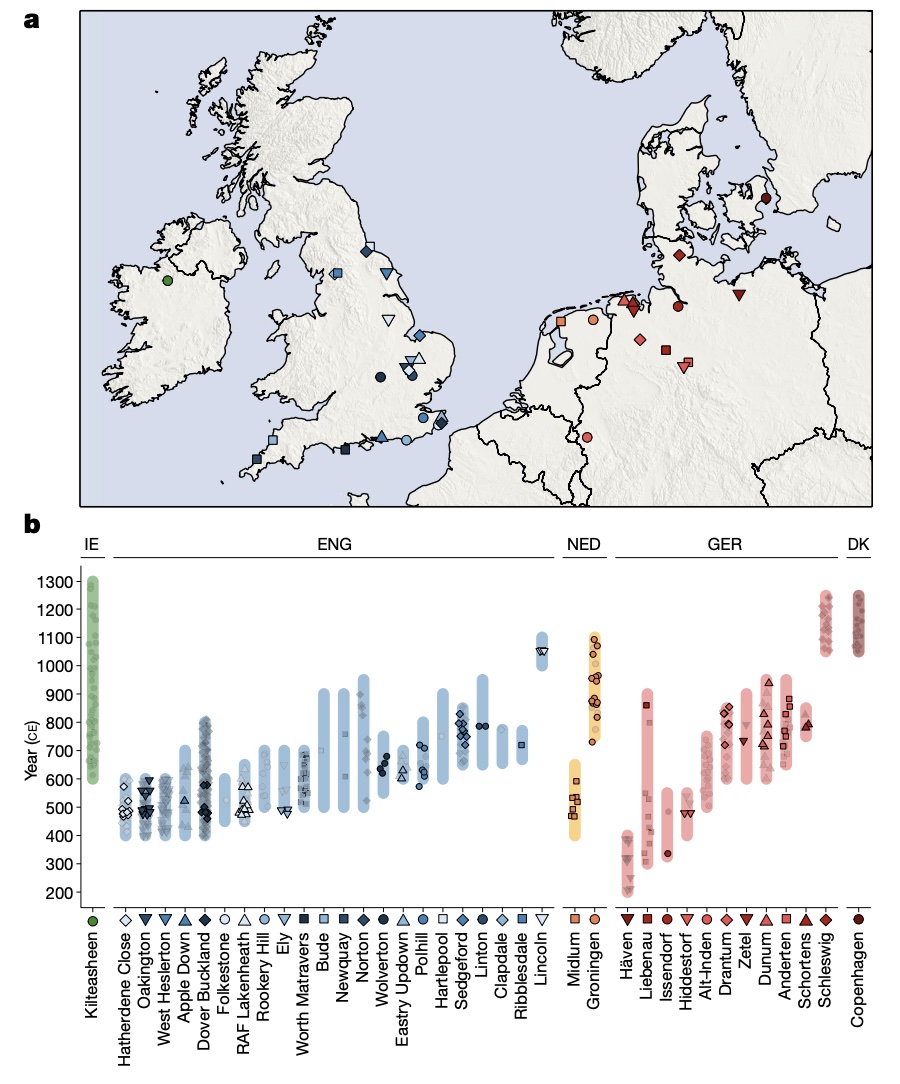
Authors: Joscha Gretzinger, Duncan Sayer, Pierre Justeau, Eveline Altena, Maria Pala, Katharina Dulias, Ceiridwen J Edwards, Susanne Jodoin, Laura Lacher, Susanna Sabin, Åshild J Vågene, Wolfgang Haak, S Sunna Ebenesersdóttir, Kristjan H S Moore, Rita Radzeviciute, Kara Schmidt, Selina Brace, Martina Abenhus Bager, Nick Patterson, Luka Papac, Nasreen Broomandkhoshbacht, Kimberly Callan, Éadaoin Harney, Lora Iliev, Ann Marie Lawson, Megan Michel, Kristin Stewardson, Fatma Zalzala, Nadin Rohland, Stefanie Kappelhoff-Beckmann, Frank Both, Daniel Winger, Daniel Neumann, Lars Saalow, Stefan Krabath, Sophie Beckett, Melanie Van Twest, Neil Faulkner, Chris Read, Tabatha Barton, Joanna Caruth, John Hines, Ben Krause-Kyora, Ursula Warnke, Verena J Schuenemann, Ian Barnes, Hanna Dahlström, Jane Jark Clausen, Andrew Richardson, Elizabeth Popescu, Natasha Dodwell, Stuart Ladd, Tom Phillips, Richard Mortimer, Faye Sayer, Diana Swales, Allison Stewart, Dominic Powlesland, Robert Kenyon, Lilian Ladle, Christina Peek, Silke Grefen-Peters, Paola Ponce, Robin Daniels, Cecily Spall, Jennifer Woolcock, Andy M Jones, Amy V Roberts, Robert Symmons, Anooshka C Rawden, Alan Cooper, Kirsten I Bos, Tom Booth, Hannes Schroeder, Mark G Thomas, Agnar Helgason, Martin B Richards, David Reich, Johannes Krause and Stephan Schiffels
Abstract:
The history of the British Isles and Ireland is characterized by multiple periods of major cultural change, including the influential transformation after the end of Roman rule, which precipitated shifts in language, settlement patterns and material culture1. The extent to which migration from continental Europe mediated these transitions is a matter of long-standing debate2–4. Here we study genome-wide ancient DNA from 460 medieval northwestern Europeans—including 278 individuals from England—alongside archaeological data, to infer contemporary population dynamics. We identify a substantial increase of continental northern European ancestry in early medieval England, which is closely related to the early medieval and present-day inhabitants of Germany and Denmark, implying large-scale substantial migration across the North Sea into Britain during the Early Middle Ages. As a result, the individuals who we analysed from eastern England derived up to 76\% of their ancestry from the continental North Sea zone, albeit with substantial regional variation and heterogeneity within sites. We show that women with immigrant ancestry were more often furnished with grave goods than women with local ancestry, whereas men with weapons were as likely not to be of immigrant ancestry. A comparison with present-day Britain indicates that subsequent demographic events reduced the fraction of continental northern European ancestry while introducing further ancestry components into the English gene pool, including substantial southwestern European ancestry most closely related to that seen in Iron Age France5,6. Archaeogenetic study of ancient DNA from medieval northwestern Europeans reveals substantial increase of continental northern European ancestry in Britain, suggesting mass migration across the North Sea during the Early Middle Ages.
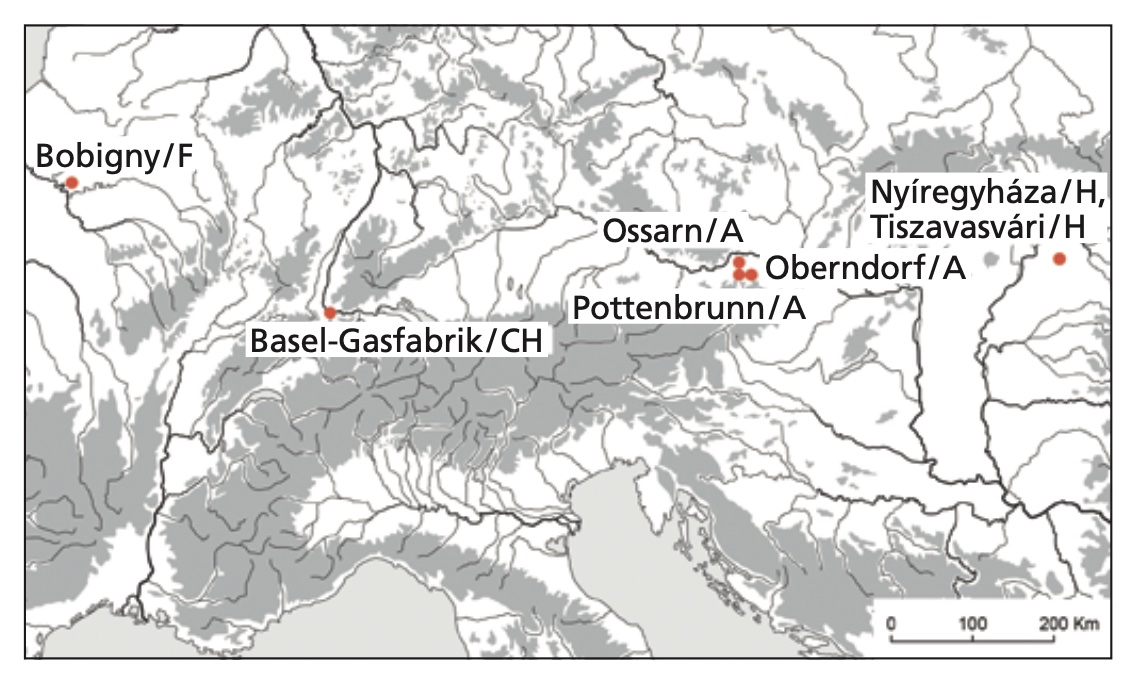
Authors: Ole Warnberg, Corina Knipper, Brigitte Roeder, Guido Lassau, Nobert Spichtig, Peter C Ramsl, Friederike Novotny, Maria Teschler-Nicola, Martin Schönfelder, Christopher F E Pare, Anna Szécsényi-Nagy, Jörg Schibler, Stephan Schiffels, Kurt W Alt and Sandra L Pichler
Abstract:
Being able to digest milk sugar beyond the age of weaning is a rather new trait in humans. The calculated age of the responsible mutations largely coincides with the introduction of dairy farming. Recent European populations exhibit a gradient of high levels of lactose tolerance in the north and lower numbers in the south. Lactase persistence is believed to have co-evolved with farming or livestock keeping as a selective advantage. Palaeogenetic data of prehistoric individuals, however, have so far not provided any clear evidence that the spread of the lactase persistence mutation predates the Roman period, while persistence increases throughout the Middle Ages. In contrast, evidence of dairy processing reaches back to the introduction of farming in the Neolithic. In this paper, we investigate lactase persistence in the La Tène period of the European Iron Age. 39 individuals from Austria, France, Hungary and Switzerland have been successfully genotyped for the two single nucleotide polymorphisms (SNPs) 13910C/T and 22018G/A, which are associated with lactose tolerance. None of those individuals carries the homozygous variant of either of the two SNPs, while four individuals are heterozygous at 22018G/A. This implies that during the Iron Age processed dairy products like cheese and yoghurt still represented the common supply of milk-derived nutrients while fresh milk played only a minor role in the regions studied here. The population-wide spread of the lactose tolerance trait in Europe therefore clearly post-dates the Iron Age.

Authors: Songül Alpaslan-Roodenberg, David Anthony, Hiba Babiker, Eszter Bánffy, Thomas Booth, Patricia Capone, Arati Deshpande-Mukherjee, Stefanie Eisenmann, Lars Fehren-Schmitz, Michael Frachetti, Ricardo Fujita, Catherine J Frieman, Qiaomei Fu, Victoria Gibbon, Wolfgang Haak, Mateja Hajdinjak, Kerstin P Hofmann, Brian Holguin, Takeshi Inomata, Hideaki Kanzawa-Kiriyama, William Keegan, Janet Kelso, Johannes Krause, Ganesan Kumaresan, Chapurukha Kusimba, Sibel Kusimba, Carles Lalueza-Fox, Bastien Llamas, Scott MacEachern, Swapan Mallick, Hirofumi Matsumura, Ana Y Morales-Arce, Giedre Motuzaite Matuzeviciute, Veena Mushrif-Tripathy, Nathan Nakatsuka, Rodrigo Nores, Christine Ogola, Mercedes Okumura, Nick Patterson, Ron Pinhasi, Samayamantri P R Prasad, Mary E Prendergast, Jose Luis Punzo, David Reich, Rikai Sawafuji, Elizabeth Sawchuk, Stephan Schiffels, Jakob Sedig, Svetlana Shnaider, Kendra Sirak, Pontus Skoglund, Viviane Slon, Meradeth Snow, Marie Soressi, Matthew Spriggs, Philipp W Stockhammer, Anna Szécsényi-Nagy, Kumarasamy Thangaraj, Vera Tiesler, Ray Tobler, Chuan-Chao Wang, Christina Warinner, Surangi Yasawardene and Muhammad Zahir
Abstract:
We are a group of archaeologists, anthropologists, curators and geneticists representing diverse global communities and 31 countries. All of us met in a virtual workshop dedicated to ethics in ancient DNA research held in November 2020. There was widespread agreement that globally applicable ethical guidelines are needed, but that recent recommendations grounded in discussion about research on human remains from North America are not always generalizable worldwide. Here we propose the following globally applicable guidelines, taking into consideration diverse contexts. These hold that: (1) researchers must ensure that all regulations were followed in the places where they work and from which the human remains derived; (2) researchers must prepare a detailed plan prior to beginning any study; (3) researchers must minimize damage to human remains; (4) researchers must ensure that data are made available following publication to allow critical re-examination of scientific findings; and (5) researchers must engage with other stakeholders from the beginning of a study and ensure respect and sensitivity to stakeholder perspectives. We commit to adhering to these guidelines and expect they will promote a high ethical standard in DNA research on human remains going forward.
Authors: Arthur Kocher, Luka Papac, Rodrigo Barquera, Felix M Key, Maria A Spyrou, Ron Hübler, Adam B Rohrlach, Franziska Aron, Raphaela Stahl, Antje Wissgott, Florian van Bömmel, Maria Pfefferkorn, Alissa Mittnik, Vanessa Villalba-Mouco, Gunnar U Neumann, Maïté Rivollat, Marieke S van de Loosdrecht, Kerttu Majander, Rezeda I Tukhbatova, Lyazzat Musralina, Ayshin Ghalichi, Sandra Penske, Susanna Sabin, Megan Michel, Joscha Gretzinger, Elizabeth A Nelson, Tiago Ferraz, Kathrin Nägele, Cody Parker, Marcel Keller, Evelyn K Guevara, Michal Feldman, Stefanie Eisenmann, Eirini Skourtanioti, Karen Giffin, Guido Alberto Gnecchi-Ruscone, Susanne Friederich, Vittoria Schimmenti, Valery Khartanovich, Marina K Karapetian, Mikhail S Chaplygin, Vladimir V Kufterin, Aleksandr A Khokhlov, Andrey A Chizhevsky, Dmitry A Stashenkov, Anna F Kochkina, Cristina Tejedor-Rodríguez, Íñigo García-Martínez de Lagrán, Héctor Arcusa-Magallón, Rafael Garrido-Pena, José Ignacio Royo-Guillén, Jan Nováček, Stéphane Rottier, Sacha Kacki, Sylvie Saintot, Elena Kaverzneva, Andrej B Belinskiy, Petr Velemínský, Petr Limburský, Michal Kostka, Louise Loe, Elizabeth Popescu, Rachel Clarke, Alice Lyons, Richard Mortimer, Antti Sajantila, Yadira Chinique de Armas, Silvia Teresita Hernandez Godoy, Diana I Hernández-Zaragoza, Jessica Pearson, Didier Binder, Philippe Lefranc, Anatoly R Kantorovich, Vladimir E Maslov, Luca Lai, Magdalena Zoledziewska, Jessica F Beckett, Michaela Langová, Alžběta Danielisová, Tara Ingman, Gabriel García Atiénzar, Maria Paz de Miguel Ibáñez, Alejandro Romero, Alessandra Sperduti, Sophie Beckett, Susannah J Salter, Emma D Zilivinskaya, Dmitry V Vasil'ev, Kristin von Heyking, Richard L Burger, Lucy C Salazar, Luc Amkreutz, Masnav Navruzbekov, Eva Rosenstock, Carmen Alonso-Fernández, Vladimir Slavchev, Alexey A Kalmykov, Biaslan Ch Atabiev, Elena Batieva, Micaela Alvarez Calmet, Bastien Llamas, Michael Schultz, Raiko Krauß, Javier Jiménez-Echevarría, Michael Francken, Svetlana Shnaider, Peter de Knijff, Eveline Altena, Katrien Van de Vijver, Lars Fehren-Schmitz, Tiffiny A Tung, Sandra Lösch, Maria Dobrovolskaya, Nikolaj Makarov, Chris Read, Melanie Van Twest, Claudia Sagona, Peter C Ramsl, Murat Akar, K Aslihan Yener, Eduardo Carmona Ballestero, Francesco Cucca, Vittorio Mazzarello, Pilar Utrilla, Kurt Rademaker, Eva Fernández-Domínguez, Douglas Baird, Patrick Semal, Lourdes Márquez-Morfín, Mirjana Roksandic, Hubert Steiner, Domingo Carlos Salazar-García, Natalia Shishlina, Yilmaz Selim Erdal, Fredrik Hallgren, Yavor Boyadzhiev, Kamen Boyadzhiev, Mario Küßner, Duncan Sayer, Päivi Onkamo, Robin Skeates, Manuel Rojo-Guerra, Alexandra Buzhilova, Elmira Khussainova, Leyla B Djansugurova, Arman Z Beisenov, Zainolla Samashev, Ken Massy, Marcello Mannino, Vyacheslav Moiseyev, Kristiina Mannermaa, Oleg Balanovsky, Marie-France Deguilloux, Sabine Reinhold, Svend Hansen, Egor P Kitov, Miroslav Dobeš, Michal Ernée, Harald Meller, Kurt W Alt, Kay Prüfer, Christina Warinner, Stephan Schiffels, Philipp W Stockhammer, Kirsten Bos, Cosimo Posth, Alexander Herbig, Wolfgang Haak, Johannes Krause and Denise Kühnert
Abstract:
[Figure: see text].
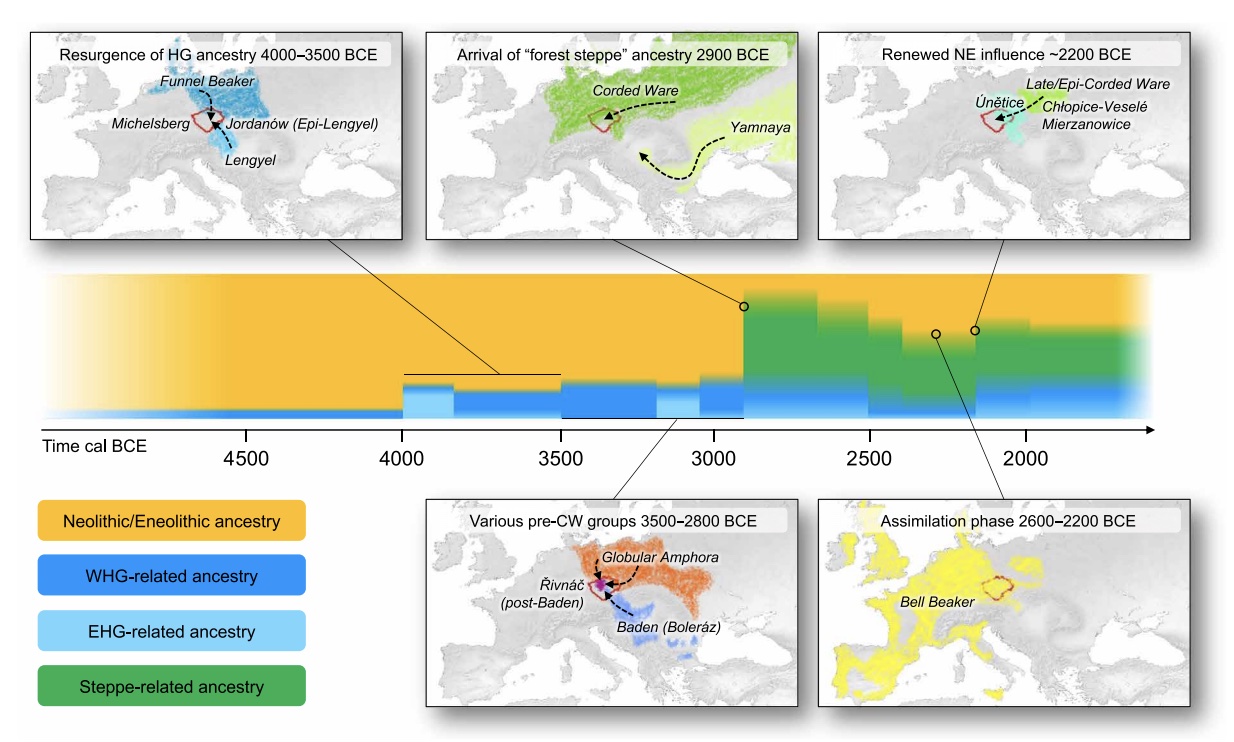
Authors: Luka Papac, Michal Ernée, Miroslav Dobeš, Michaela Langová, Adam B Rohrlach, Franziska Aron, Gunnar U Neumann, Maria A Spyrou, Nadin Rohland, Petr Velemínský, Martin Kuna, Hana Brzobohatá, Brendan Culleton, David Daněček, Alžběta Danielisová, Miluše Dobisíková, Josef Hložek, Douglas J Kennett, Jana Klementová, Michal Kostka, Petr Krištuf, Milan Kuchařík, Jana Kuljavceva Hlavová, Petr Limburský, Drahomíra Malyková, Lucia Mattiello, Monika Pecinovská, Katarína Petriščáková, Erika Průchová, Petra Stránská, Lubor Smejtek, Jaroslav Špaček, Radka Šumberová, Ondřej Švejcar, Martin Trefný, Miloš Vávra, Jan Kolář, Volker Heyd, Johannes Krause, Ron Pinhasi, David Reich, Stephan Schiffels and Wolfgang Haak
Abstract:
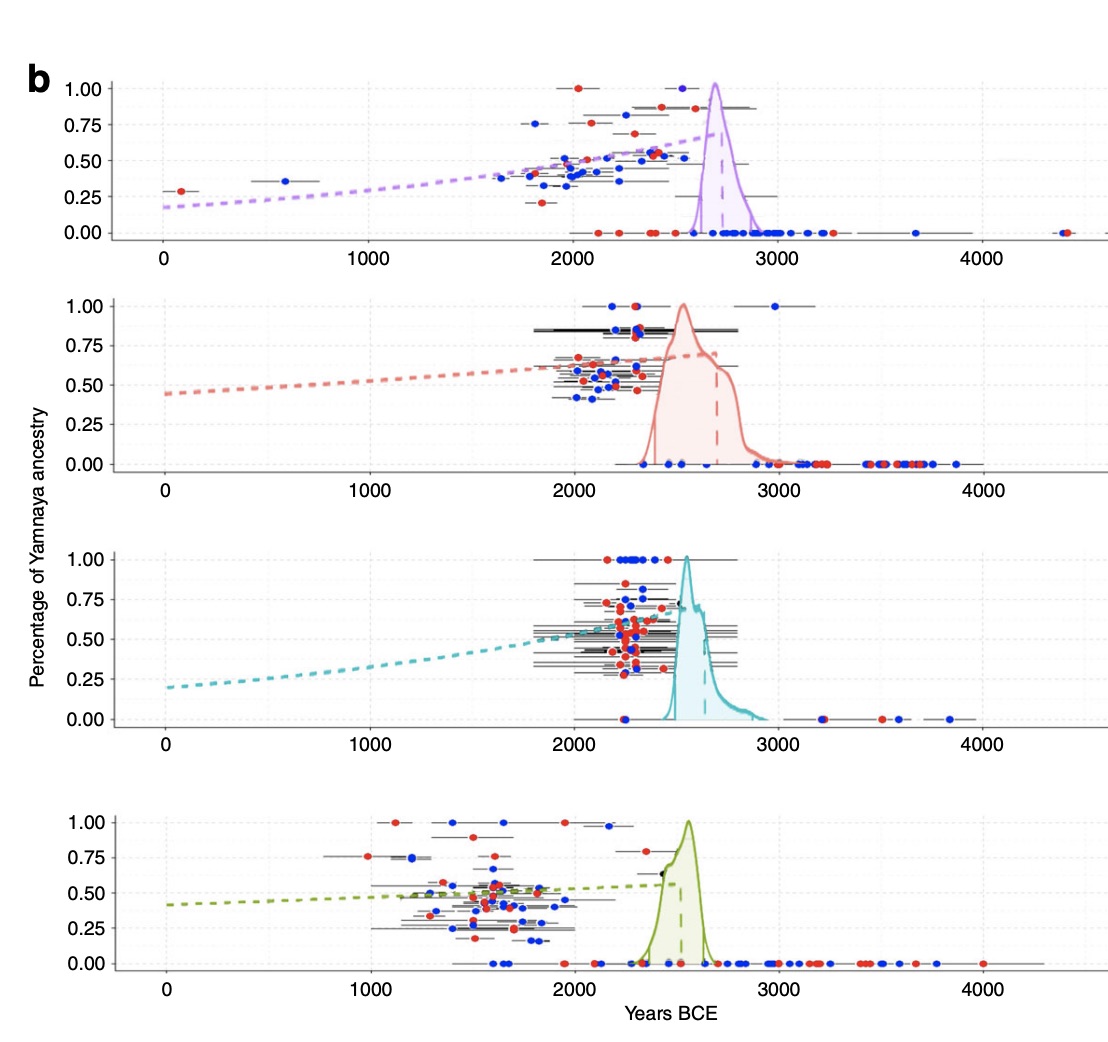
Europe's prehistory oversaw dynamic and complex interactions of diverse societies, hitherto unexplored at detailed regional scales. Studying 271 human genomes dated ~4900 to 1600 BCE from the European heartland, Bohemia, we reveal unprecedented genetic changes and social processes. Major migrations preceded the arrival of ``steppe'' ancestry, and at ~2800 BCE, three genetically and culturally differentiated groups coexisted. Corded Ware appeared by 2900 BCE, were initially genetically diverse, did not derive all steppe ancestry from known Yamnaya, and assimilated females of diverse backgrounds. Both Corded Ware and Bell Beaker groups underwent dynamic changes, involving sharp reductions and complete replacements of Y-chromosomal diversity at ~2600 and ~2400 BCE, respectively, the latter accompanied by increased Neolithic-like ancestry. The Bronze Age saw new social organization emerge amid a ≥40\% population turnover.
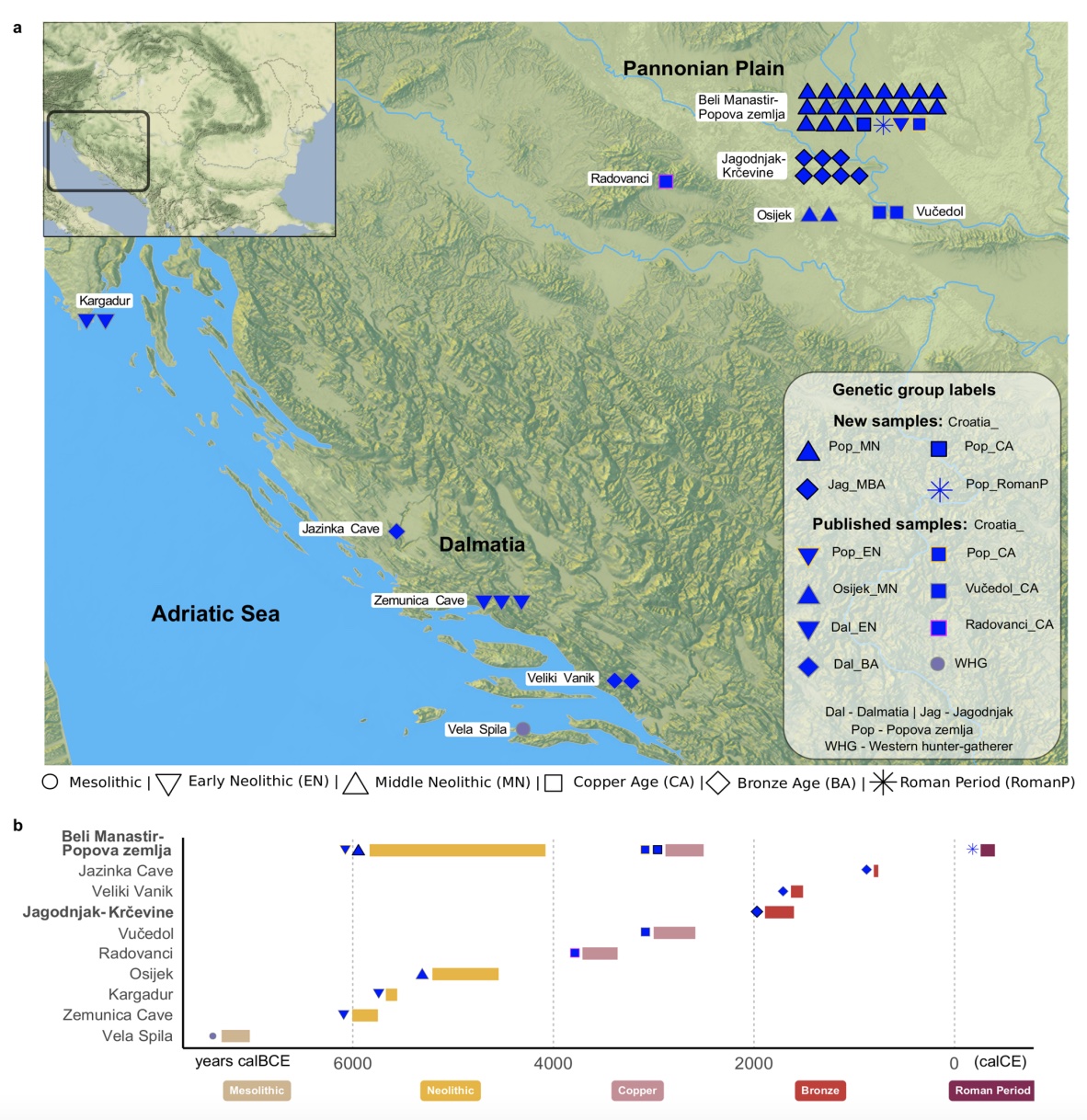
Authors: Suzanne Freilich, Harald Ringbauer, Dženi Los, Mario Novak, Dinko Tresić Pavičić, Stephan Schiffels and Ron Pinhasi
Abstract:
Ancient DNA studies have revealed how human migrations from the Neolithic to the Bronze Age transformed the social and genetic structure of European societies. Present-day Croatia lies at the heart of ancient migration routes through Europe, yet our knowledge about social and genetic processes here remains sparse. To shed light on these questions, we report new whole-genome data for 28 individuals dated to between ~ 4700 BCE–400 CE from two sites in present-day eastern Croatia. In the Middle Neolithic we evidence first cousin mating practices and strong genetic continuity from the Early Neolithic. In the Middle Bronze Age community that we studied, we find multiple closely related males suggesting a patrilocal social organisation. We also find in that community an unexpected genetic ancestry profile distinct from individuals found at contemporaneous sites in the region, due to the addition of hunter-gatherer-related ancestry. These findings support archaeological evidence for contacts with communities further north in the Carpathian Basin. Finally, an individual dated to Roman times exhibits an ancestry profile that is broadly present in the region today, adding an important data point to the substantial shift in ancestry that occurred in the region between the Bronze Age and today.
Authors: Wiebke Ahrndt, Larissa Förster, Sarah Fründt, Michael Geißdorf, Diana Gabler, Bernhard Heeb, Christian Lenk, Susanne Roeßiger, Stephan Schiffels, Carola Thielecke, Hilke Thode-Arora, Andreas Winkelmann, Edward} {Halealoha Ayau of Pana`ewa, Michael Pickering, Alma Mekondjo Nankela, Jeremy Silvester and Anne Wesche
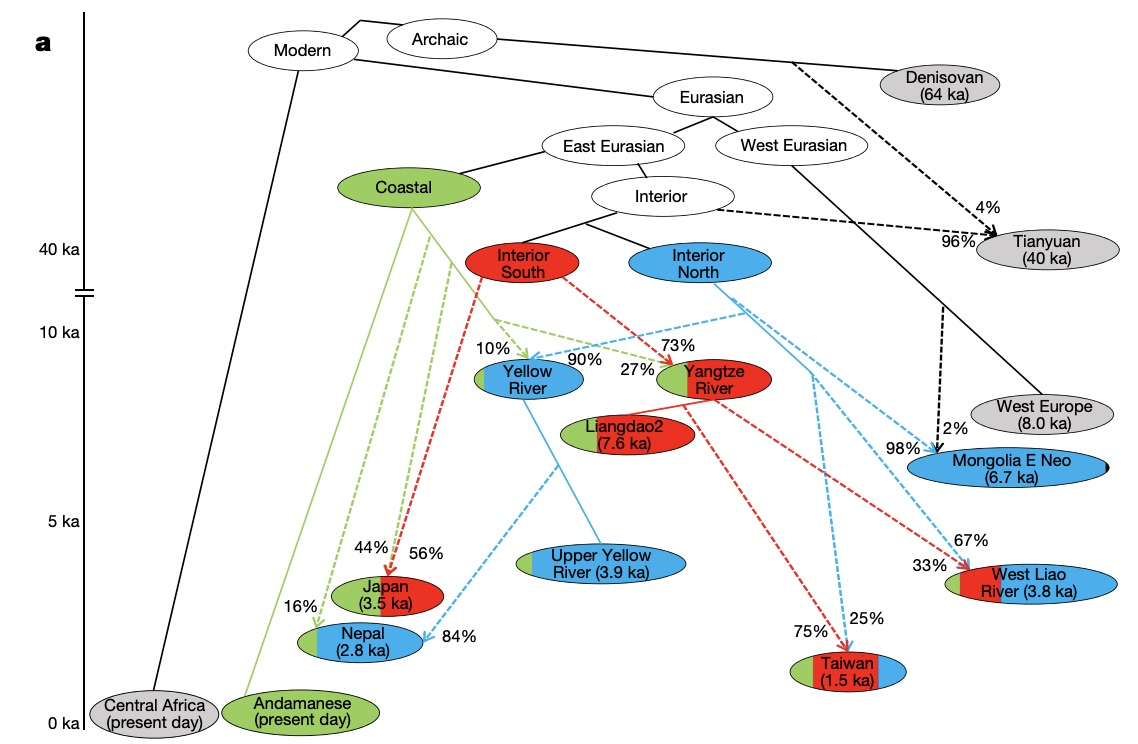
Authors: Chuan-Chao Wang, Hui-Yuan Yeh, Alexander N Popov, Hu-Qin Zhang, Hirofumi Matsumura, Kendra Sirak, Olivia Cheronet, Alexey Kovalev, Nadin Rohland, Alexander M Kim, Swapan Mallick, Rebecca Bernardos, Dashtseveg Tumen, Jing Zhao, Yi-Chang Liu, Jiun-Yu Liu, Matthew Mah, Ke Wang, Zhao Zhang, Nicole Adamski, Nasreen Broomandkhoshbacht, Kimberly Callan, Francesca Candilio, Kellie Sara Duffett Carlson, Brendan J Culleton, Laurie Eccles, Suzanne Freilich, Denise Keating, Ann Marie Lawson, Kirsten Mandl, Megan Michel, Jonas Oppenheimer, Kadir Toykan Özdoğan, Kristin Stewardson, Shaoqing Wen, Shi Yan, Fatma Zalzala, Richard Chuang, Ching-Jung Huang, Hana Looh, Chung-Ching Shiung, Yuri G Nikitin, Andrei V Tabarev, Alexey A Tishkin, Song Lin, Zhou-Yong Sun, Xiao-Ming Wu, Tie-Lin Yang, Xi Hu, Liang Chen, Hua Du, Jamsranjav Bayarsaikhan, Enkhbayar Mijiddorj, Diimaajav Erdenebaatar, Tumur-Ochir Iderkhangai, Erdene Myagmar, Hideaki Kanzawa-Kiriyama, Masato Nishino, Ken-Ichi Shinoda, Olga A Shubina, Jianxin Guo, Wangwei Cai, Qiongying Deng, Longli Kang, Dawei Li, Dongna Li, Rong Lin, Nini , Rukesh Shrestha, Ling-Xiang Wang, Lanhai Wei, Guangmao Xie, Hongbing Yao, Manfei Zhang, Guanglin He, Xiaomin Yang, Rong Hu, Martine Robbeets, Stephan Schiffels, Douglas J Kennett, Li Jin, Hui Li, Johannes Krause, Ron Pinhasi and David Reich
Abstract:
The deep population history of East Asia remains poorly understood owing to a lack of ancient DNA data and sparse sampling of present-day people1,2. Here we report genome-wide data from 166 East Asian individuals dating to between 6000 BC and AD 1000 and 46 present-day groups. Hunter-gatherers from Japan, the Amur River Basin, and people of Neolithic and Iron Age Taiwan and the Tibetan Plateau are linked by a deeply splitting lineage that probably reflects a coastal migration during the Late Pleistocene epoch. We also follow expansions during the subsequent Holocene epoch from four regions. First, hunter-gatherers from Mongolia and the Amur River Basin have ancestry shared by individuals who speak Mongolic and Tungusic languages, but do not carry ancestry characteristic of farmers from the West Liao River region (around 3000 BC), which contradicts theories that the expansion of these farmers spread the Mongolic and Tungusic proto-languages. Second, farmers from the Yellow River Basin (around 3000 BC) probably spread Sino-Tibetan languages, as their ancestry dispersed both to Tibet-where it forms approximately 84\% of the gene pool in some groups-and to the Central Plain, where it has contributed around 59-84\% to modern Han Chinese groups. Third, people from Taiwan from around 1300 BC to AD 800 derived approximately 75\% of their ancestry from a lineage that is widespread in modern individuals who speak Austronesian, Tai-Kadai and Austroasiatic languages, and that we hypothesize derives from farmers of the Yangtze River Valley. Ancient people from Taiwan also derived about 25\% of their ancestry from a northern lineage that is related to, but different from, farmers of the Yellow River Basin, which suggests an additional north-to-south expansion. Fourth, ancestry from Yamnaya Steppe pastoralists arrived in western Mongolia after around 3000 BC but was displaced by previously established lineages even while it persisted in western China, as would be expected if this ancestry was associated with the spread of proto-Tocharian Indo-European languages. Two later gene flows affected western Mongolia: migrants after around 2000 BC with Yamnaya and European farmer ancestry, and episodic influences of later groups with ancestry from Turan.
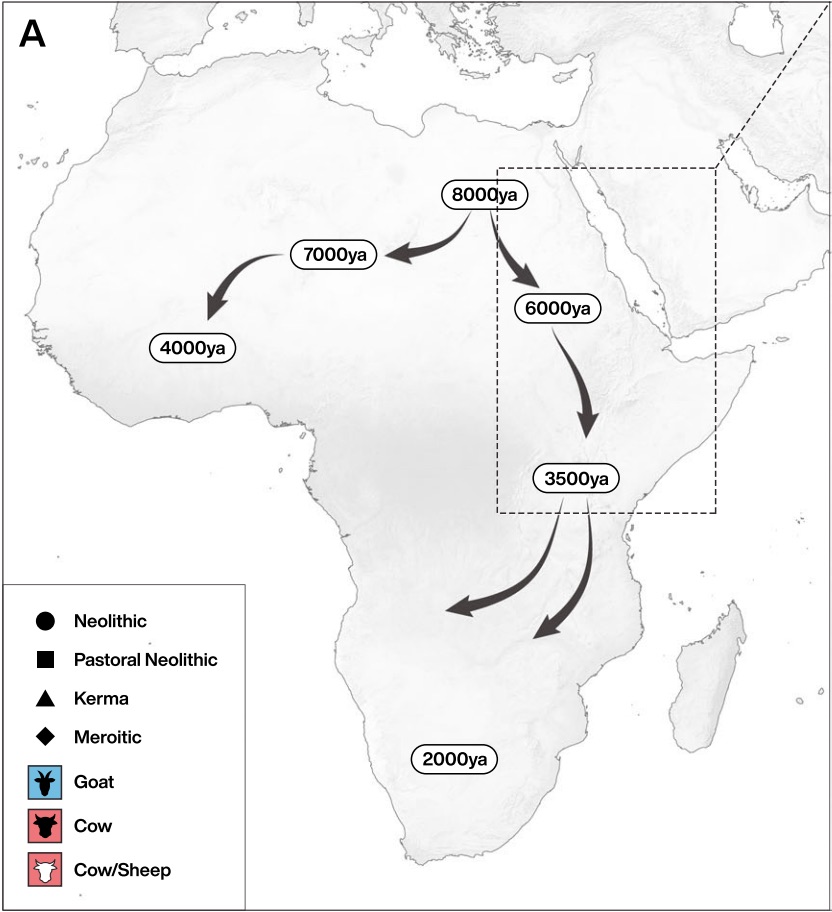
Authors: Madeleine Bleasdale, Kristine K Richter, Anneke Janzen, Samantha Brown, Ashley Scott, Jana Zech, Shevan Wilkin, Ke Wang, Stephan Schiffels, Jocelyne Desideri, Marie Besse, Jacques Reinold, Mohamed Saad, Hiba Babiker, Robert C Power, Emmanuel Ndiema, Christine Ogola, Fredrick K Manthi, Muhammad Zahir, Michael Petraglia, Christian Trachsel, Paolo Nanni, Jonas Grossmann, Jessica Hendy, Alison Crowther, Patrick Roberts, Steven T Goldstein and Nicole Boivin
Abstract:
Consuming the milk of other species is a unique adaptation of Homo sapiens, with implications for health, birth spacing and evolution. Key questions nonetheless remain regarding the origins of dairying and its relationship to the genetically-determined ability to drink milk into adulthood through lactase persistence (LP). As a major centre of LP diversity, Africa is of significant interest to the evolution of dairying. Here we report proteomic evidence for milk consumption in ancient Africa. Using liquid chromatography tandem mass spectrometry (LC-MS/MS) we identify dairy proteins in human dental calculus from northeastern Africa, directly demonstrating milk consumption at least six millennia ago. Our findings indicate that pastoralist groups were drinking milk as soon as herding spread into eastern Africa, at a time when the genetic adaptation for milk digestion was absent or rare. Our study links LP status in specific ancient individuals with direct evidence for their consumption of dairy products.
Authors: Choongwon Jeong, Ke Wang, Shevan Wilkin, William Timothy Treal Taylor, Bryan K Miller, Jan H Bemmann, Raphaela Stahl, Chelsea Chiovelli, Florian Knolle, Sodnom Ulziibayar, Dorjpurev Khatanbaatar, Diimaajav Erdenebaatar, Ulambayar Erdenebat, Ayudai Ochir, Ganbold Ankhsanaa, Chuluunkhuu Vanchigdash, Battuga Ochir, Chuluunbat Munkhbayar, Dashzeveg Tumen, Alexey Kovalev, Nikolay Kradin, Bilikto A Bazarov, Denis A Miyagashev, Prokopiy B Konovalov, Elena Zhambaltarova, Alicia Ventresca Miller, Wolfgang Haak, Stephan Schiffels, Johannes Krause, Nicole Boivin, Myagmar Erdene, Jessica Hendy and Christina Warinner
Authors: Ke Wang, Steven Goldstein, Madeleine Bleasdale, Bernard Clist, Koen Bostoen, Paul Bakwa-Lufu, Laura T Buck, Alison Crowther, Alioune Dème, Roderick J McIntosh, Julio Mercader, Christine Ogola, Robert C Power, Elizabeth Sawchuk, Peter Robertshaw, Edwin N Wilmsen, Michael Petraglia, Emmanuel Ndiema, Fredrick K Manthi, Johannes Krause, Patrick Roberts, Nicole Boivin and Stephan Schiffels
Abstract:
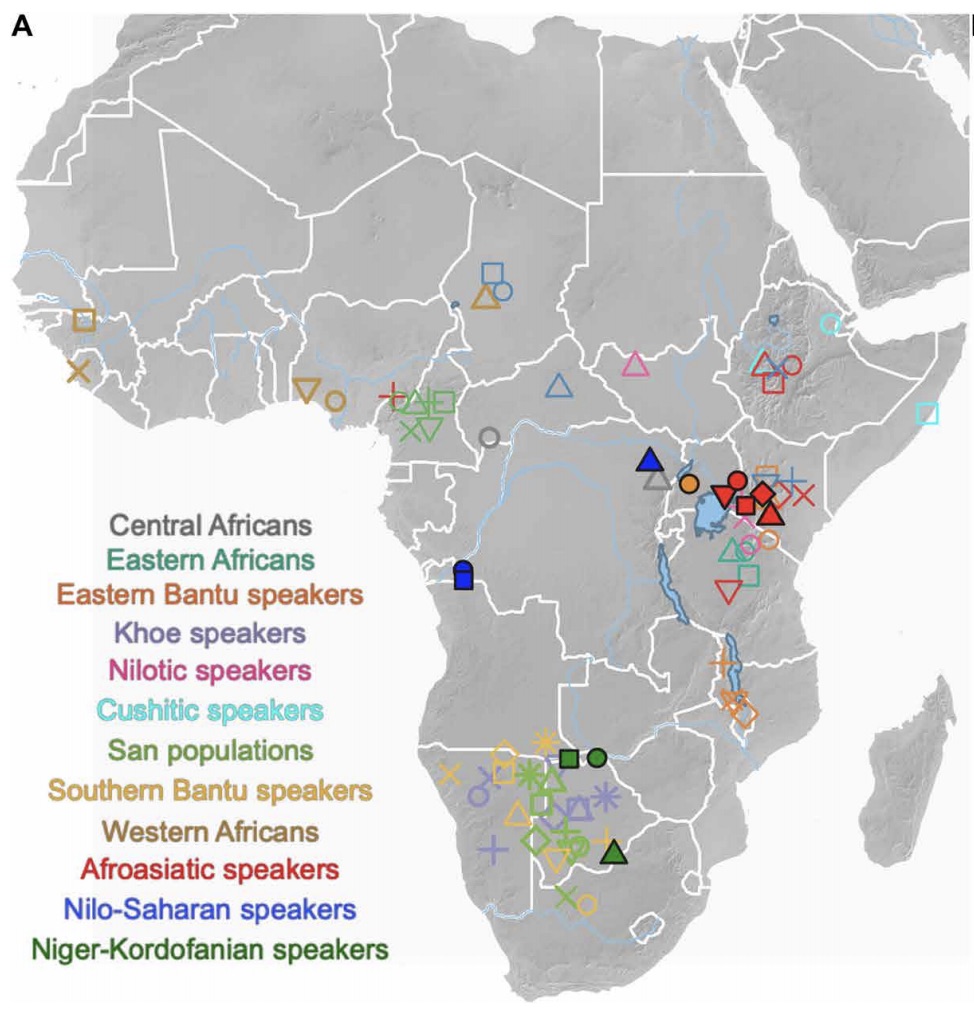
Africa hosts the greatest human genetic diversity globally, but legacies of ancient population interactions and dispersals across the continent remain understudied. Here, we report genome-wide data from 20 ancient sub-Saharan African individuals, including the first reported ancient DNA from the DRC, Uganda, and Botswana. These data demonstrate the contraction of diverse, once contiguous hunter-gatherer populations, and suggest the resistance to interaction with incoming pastoralists of delayed-return foragers in aquatic environments. We refine models for the spread of food producers into eastern and southern Africa, demonstrating more complex trajectories of admixture than previously suggested. In Botswana, we show that Bantu ancestry post-dates admixture between pastoralists and foragers, suggesting an earlier spread of pastoralism than farming to southern Africa. Our findings demonstrate how processes of migration and admixture have markedly reshaped the genetic map of sub-Saharan Africa in the past few millennia and highlight the utility of combined archaeological and archaeogenetic approaches.
Authors: Maïté Rivollat, Choongwon Jeong, Stephan Schiffels, İşil Küçükkalıpçı, Marie-Hélène Pemonge, Adam Benjamin Rohrlach, Kurt W Alt, Didier Binder, Susanne Friederich, Emmanuel Ghesquière, Detlef Gronenborn, Luc Laporte, Philippe Lefranc, Harald Meller, Hélène Réveillas, Eva Rosenstock, Stéphane Rottier, Chris Scarre, Ludovic Soler, Joachim Wahl, Johannes Krause, Marie-France Deguilloux and Wolfgang Haak
Abstract:
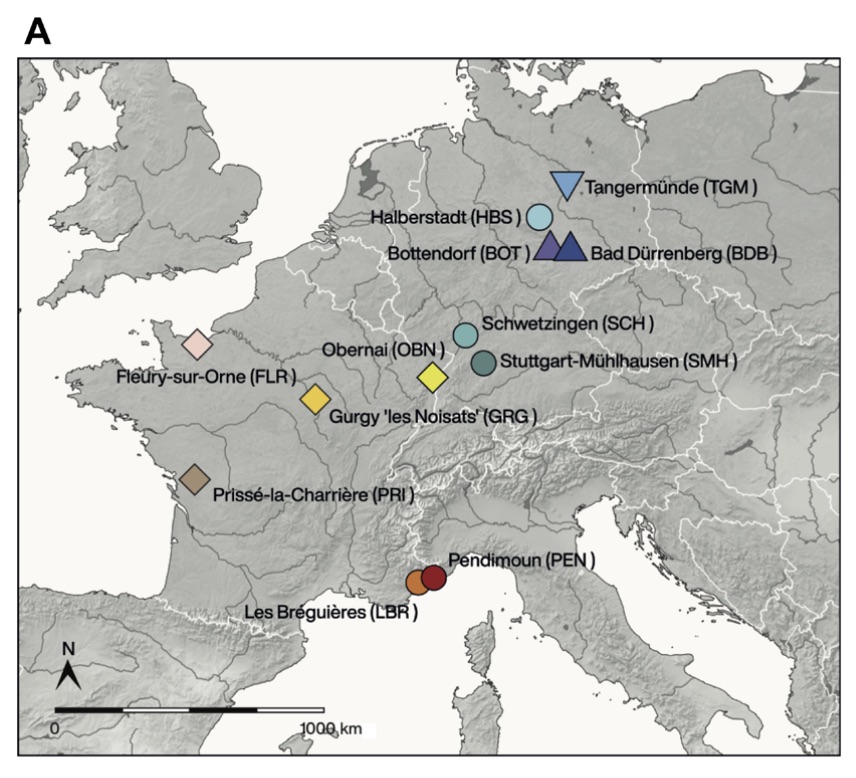
Starting from 12,000 years ago in the Middle East, the Neolithic lifestyle spread across Europe via separate continental and Mediterranean routes. Genomes from early European farmers have shown a clear Near Eastern/Anatolian genetic affinity with limited contribution from hunter-gatherers. However, no genomic data are available from modern-day France, where both routes converged, as evidenced by a mosaic cultural pattern. Here, we present genome-wide data from 101 individuals from 12 sites covering today's France and Germany from the Mesolithic (N = 3) to the Neolithic (N = 98) (7000–3000 BCE). Using the genetic substructure observed in European hunter-gatherers, we characterize diverse patterns of admixture in different regions, consistent with both routes of expansion. Early western European farmers show a higher proportion of distinctly western hunter-gatherer ancestry compared to central/southeastern farmers. Our data highlight the complexity of the biological interactions during the Neolithic expansion by revealing major regional variations.
Authors: Anja Furtwängler, A B Rohrlach, Thiseas C Lamnidis, Luka Papac, Gunnar U Neumann, Inga Siebke, Ella Reiter, Noah Steuri, Jürgen Hald, Anthony Denaire, Bernadette Schnitzler, Joachim Wahl, Marianne Ramstein, Verena J Schuenemann, Philipp W Stockhammer, Albert Hafner, Sandra Lösch, Wolfgang Haak, Stephan Schiffels and Johannes Krause
Abstract:
Genetic studies of Neolithic and Bronze Age skeletons from Europe have provided evidence for strong population genetic changes at the beginning and the end of the Neolithic period. To further understand the implications of these in Southern Central Europe, we analyze 96 ancient genomes from Switzerland, Southern Germany, and the Alsace region in France, covering the Middle/Late Neolithic to Early Bronze Age. Similar to previously described genetic changes in other parts of Europe from the early 3rd millennium BCE, we detect an arrival of ancestry related to Late Neolithic pastoralists from the Pontic-Caspian steppe in Switzerland as early as 2860–2460 calBCE. Our analyses suggest that this genetic turnover was a complex process lasting almost 1000 years and involved highly genetically structured populations in this region. European populations underwent strong genetic changes during the Neolithic. Here, Furtwängler et al. provide ancient nuclear and mitochondrial genomic data from the region of Switzerland during the end of the Neolithic and the Early Bronze Age that reveal a complex genetic turnover during the arrival of steppe ancestry.
Authors: Marieke S van de Loosdrecht, Marcello A Mannino, Sahra Talamo, Vanessa Villalba-Mouco, Cosimo Posth, Franziska Aron, Guido Brandt, Marta Burri, Cäcilia Freund, Rita Radzeviciute, Raphaela Stahl, Antje Wissgott, Lysann Klausnitzer, Sarah Nagel, Matthias Meyer, Antonio Tagliacozzo, Marcello Piperno, Sebastiano Tusa, Carmine Collina, Vittoria Schimmenti, Rosaria Di Salvo, Kay Prüfer, Jean-Jacques Hublin, Stephan Schiffels, Choongwon Jeong, Wolfgang Haak and Johannes Krause
Abstract:
Abstract Southern Italy is a key region for understanding the agricultural transition in the Mediterranean due to its central position. We present a genomic transect for 19 prehistoric Sicilians that covers the Early Mesolithic to Early Neolithic period. We find that the Early Mesolithic hunter-gatherers (HGs) are a highly drifted sister lineage to Early Holocene western European HGs, whereas a quarter of the Late Mesolithic HGs ancestry is related to HGs from eastern Europe and the Near East. This indicates substantial gene flow from (south-)eastern Europe between the Early and Late Mesolithic. The Early Neolithic farmers are genetically most similar to those from the Balkan and Greece, and carry only a maximum of ∼7\% ancestry from Sicilian Mesolithic HGs. Ancestry changes match changes in dietary profile and material culture, except for two individuals who may provide tentative initial evidence that HGs adopted elements of farming in Sicily.One-sentence summary Genome-wide and isotopic data from prehistoric Sicilians reveal a pre-farming connection to (south-) eastern Europe, and tentative initial evidence that hunter-gatherers adopted some Neolithic aspects prior to near-total replacement by early farmers.
Authors: Ke Wang, Iain Mathieson, Jared O'Connell and Stephan Schiffels
Abstract:
The genetic diversity of humans, like many species, has been shaped by a complex pattern of population separations followed by isolation and subsequent admixture. This pattern, reaching at least as far back as the appearance of our species in the paleontological record, has left its traces in our genomes. Reconstructing a population's history from these traces is a challenging problem. Here we present a novel approach based on the Multiple Sequentially Markovian Coalescent (MSMC) to analyze the separation history between populations. Our approach, called MSMC-IM, uses an improved implementation of the MSMC (MSMC2) to estimate coalescence rates within and across pairs of populations, and then fits a continuous Isolation-Migration model to these rates to obtain a time-dependent estimate of gene flow. We show, using simulations, that our method can identify complex demographic scenarios involving post-split admixture or archaic introgression. We apply MSMC-IM to whole genome sequences from 15 worldwide populations, tracking the process of human genetic diversification. We detect traces of extremely deep ancestry between some African populations, with around 1\% of ancestry dating to divergences older than a million years ago.
Authors: Stephan Schiffels and Ke Wang
Abstract:
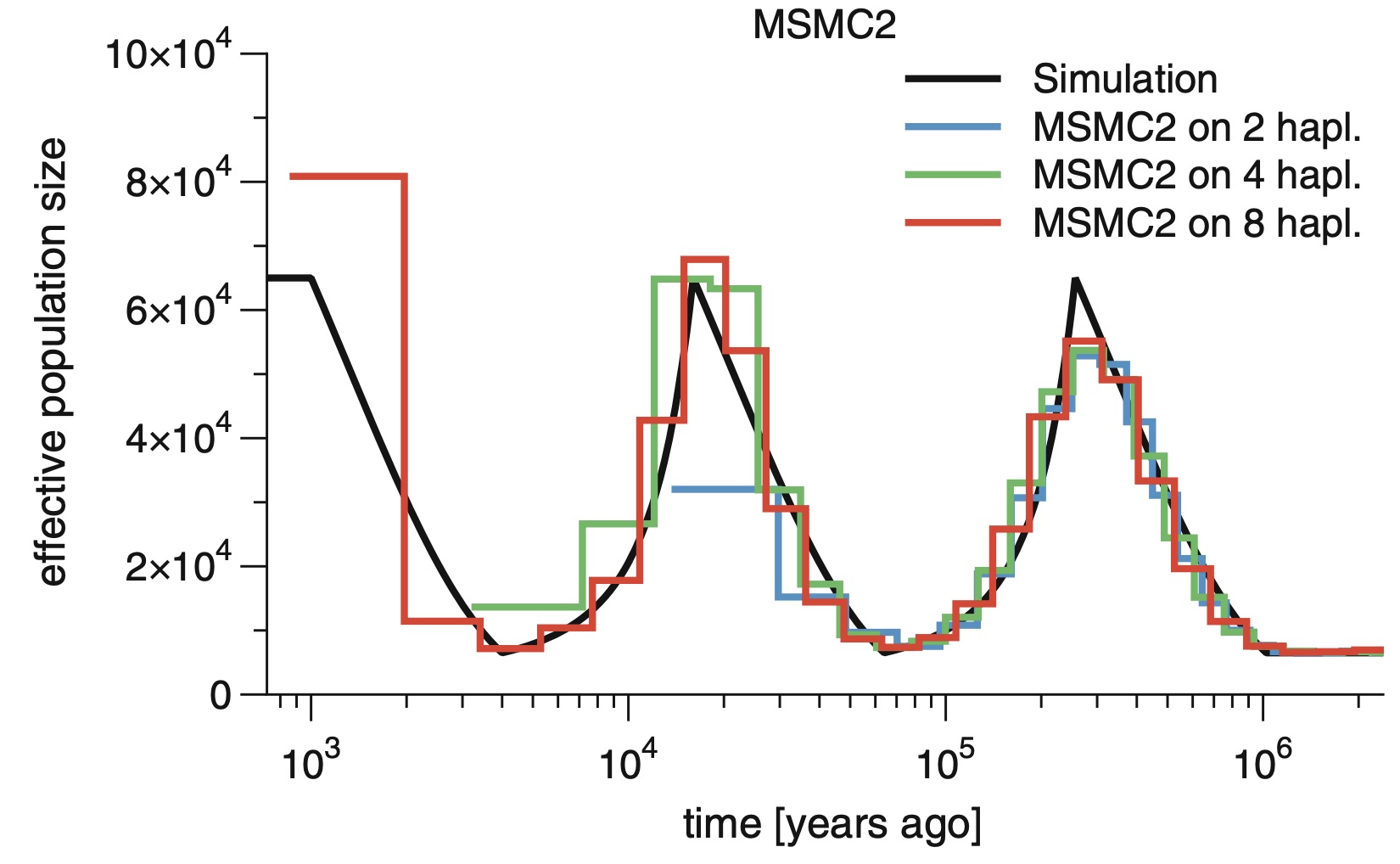
The Multiple Sequentially Markovian Coalescent (MSMC) is a population genetic method and software for inferring demographic history and population structure through time from genome sequences. Here we describe the main program MSMC and its successor MSMC2. We go through all the necessary steps of processing genomic data from BAM files all the way to generating plots of inferred population size and separation histories. Some background on the methodology itself is provided, as well as bash scripts and python source code to run the necessary programs. The reader is also referred to community resources such as a mailing list and github repositories for further advice.
Authors: Alissa Mittnik, Ken Massy, Corina Knipper, Fabian Wittenborn, Ronny Friedrich, Saskia Pfrengle, Marta Burri, Nadine Carlichi-Witjes, Heidi Deeg, Anja Furtwängler, Michaela Harbeck, Kristin von Heyking, Catharina Kociumaka, Isil Kucukkalipci, Susanne Lindauer, Stephanie Metz, Anja Staskiewicz, Andreas Thiel, Joachim Wahl, Wolfgang Haak, Ernst Pernicka, Stephan Schiffels, Philipp W Stockhammer and Johannes Krause
Abstract:
Revealing and understanding the mechanisms behind social inequality in prehistoric societies is a major challenge. By combining genome-wide data, isotopic evidence, and anthropological and archaeological data, we have gone beyond the dominating supraregional approaches in archaeogenetics to shed light on the complexity of social status, inheritance rules, and mobility during the Bronze Age. We applied a deep microregional approach and analyzed genome-wide data of 104 human individuals deriving from farmstead-related cemeteries from the Late Neolithic to the Middle Bronze Age in southern Germany. Our results reveal individual households, lasting several generations, that consisted of a high-status core family and unrelated low-status individuals; a social organization accompanied by patrilocality and female exogamy; and the stability of this system over 700 years.
Authors: Deepti Gurdasani, Tommy Carstensen, Segun Fatumo, Guanjie Chen, Chris S Franklin, Javier Prado-Martinez, Heleen Bouman, Federico Abascal, Marc Haber, Ioanna Tachmazidou, Iain Mathieson, Kenneth Ekoru, Marianne K DeGorter, Rebecca N Nsubuga, Chris Finan, Eleanor Wheeler, Li Chen, David N Cooper, Stephan Schiffels, Yuan Chen, Graham R S Ritchie, Martin O Pollard, Mary D Fortune, Alex J Mentzer, Erik Garrison, Anders Bergström, Konstantinos Hatzikotoulas, Adebowale Adeyemo, Ayo Doumatey, Heather Elding, Louise V Wain, George Ehret, Paul L Auer, Charles L Kooperberg, Alexander P Reiner, Nora Franceschini, Dermot P Maher, Stephen B Montgomery, Carl Kadie, Chris Widmer, Yali Xue, Janet Seeley, Gershim Asiki, Anatoli Kamali, Elizabeth H Young, Cristina Pomilla, Nicole Soranzo, Eleftheria Zeggini, Fraser Pirie, Andrew P Morris, David Heckerman, Chris Tyler-Smith, Ayesha Motala, Charles Rotimi, Pontiano Kaleebu, Ines Barroso and Manj S Sandhu
Abstract:
Genomic studies in African populations provide unique opportunities to understand disease etiology, human diversity, and population history. In the largest study of its kind, comprising genome-wide data from 6,400 individuals and whole-genome sequences from 1,978 individuals from rural Uganda, we find evidence of geographically correlated fine-scale population substructure. Historically, the ancestry of modern Ugandans was best represented by a mixture of ancient East African pastoralists. We demonstrate the value of the largest sequence panel from Africa to date as an imputation resource. Examining 34 cardiometabolic traits, we show systematic differences in trait heritability between European and African populations, probably reflecting the differential impact of genes and environment. In a multi-trait pan-African GWAS of up to 14,126 individuals, we identify novel loci associated with anthropometric, hematological, lipid, and glycemic traits. We find that several functionally important signals are driven by Africa-specific variants, highlighting the value of studying diverse populations across the region.

Authors: Pavel Flegontov, N Ezgi Altınışık, Piya Changmai, Nadin Rohland, Swapan Mallick, Nicole Adamski, Deborah A Bolnick, Nasreen Broomandkhoshbacht, Francesca Candilio, Brendan J Culleton, Olga Flegontova, T Max Friesen, Choongwon Jeong, Thomas K Harper, Denise Keating, Douglas J Kennett, Alexander M Kim, Thiseas C Lamnidis, Ann Marie Lawson, Iñigo Olalde, Jonas Oppenheimer, Ben A Potter, Jennifer Raff, Robert A Sattler, Pontus Skoglund, Kristin Stewardson, Edward J Vajda, Sergey Vasilyev, Elizaveta Veselovskaya, M Geoffrey Hayes, Dennis H O'Rourke, Johannes Krause, Ron Pinhasi, David Reich and Stephan Schiffels
Abstract:
Much of the American Arctic was first settled 5,000 years ago, by groups of people known as Palaeo-Eskimos. They were subsequently joined and largely displaced around 1,000 years ago by ancestors of the present-day Inuit and Yup'ik1-3. The genetic relationship between Palaeo-Eskimos and Native American, Inuit, Yup'ik and Aleut populations remains uncertain4-6. Here we present genomic data for 48 ancient individuals from Chukotka, East Siberia, the Aleutian Islands, Alaska, and the Canadian Arctic. We co-analyse these data with data from present-day Alaskan Iñupiat and West Siberian populations and published genomes. Using methods based on rare-allele and haplotype sharing, as well as established techniques4,7-9, we show that Palaeo-Eskimo-related ancestry is ubiquitous among people who speak Na-Dene and Eskimo-Aleut languages. We develop a comprehensive model for the Holocene peopling events of Chukotka and North America, and show that Na-Dene-speaking peoples, people of the Aleutian Islands, and Yup'ik and Inuit across the Arctic region all share ancestry from a single Palaeo-Eskimo-related Siberian source.
Authors: Choongwon Jeong, Oleg Balanovsky, Elena Lukianova, Nurzhibek Kahbatkyzy, Pavel Flegontov, Valery Zaporozhchenko, Alexander Immel, Chuan-Chao Wang, Olzhas Ixan, Elmira Khussainova, Bakhytzhan Bekmanov, Victor Zaibert, Maria Lavryashina, Elvira Pocheshkhova, Yuldash Yusupov, Anastasiya Agdzhoyan, Sergey Koshel, Andrei Bukin, Pagbajabyn Nymadawa, Shahlo Turdikulova, Dilbar Dalimova, Mikhail Churnosov, Roza Skhalyakho, Denis Daragan, Yuri Bogunov, Anna Bogunova, Alexandr Shtrunov, Nadezhda Dubova, Maxat Zhabagin, Levon Yepiskoposyan, Vladimir Churakov, Nikolay Pislegin, Larissa Damba, Ludmila Saroyants, Khadizhat Dibirova, Lubov Atramentova, Olga Utevska, Eldar Idrisov, Evgeniya Kamenshchikova, Irina Evseeva, Mait Metspalu, Alan K Outram, Martine Robbeets, Leyla Djansugurova, Elena Balanovska, Stephan Schiffels, Wolfgang Haak, David Reich and Johannes Krause
Abstract:
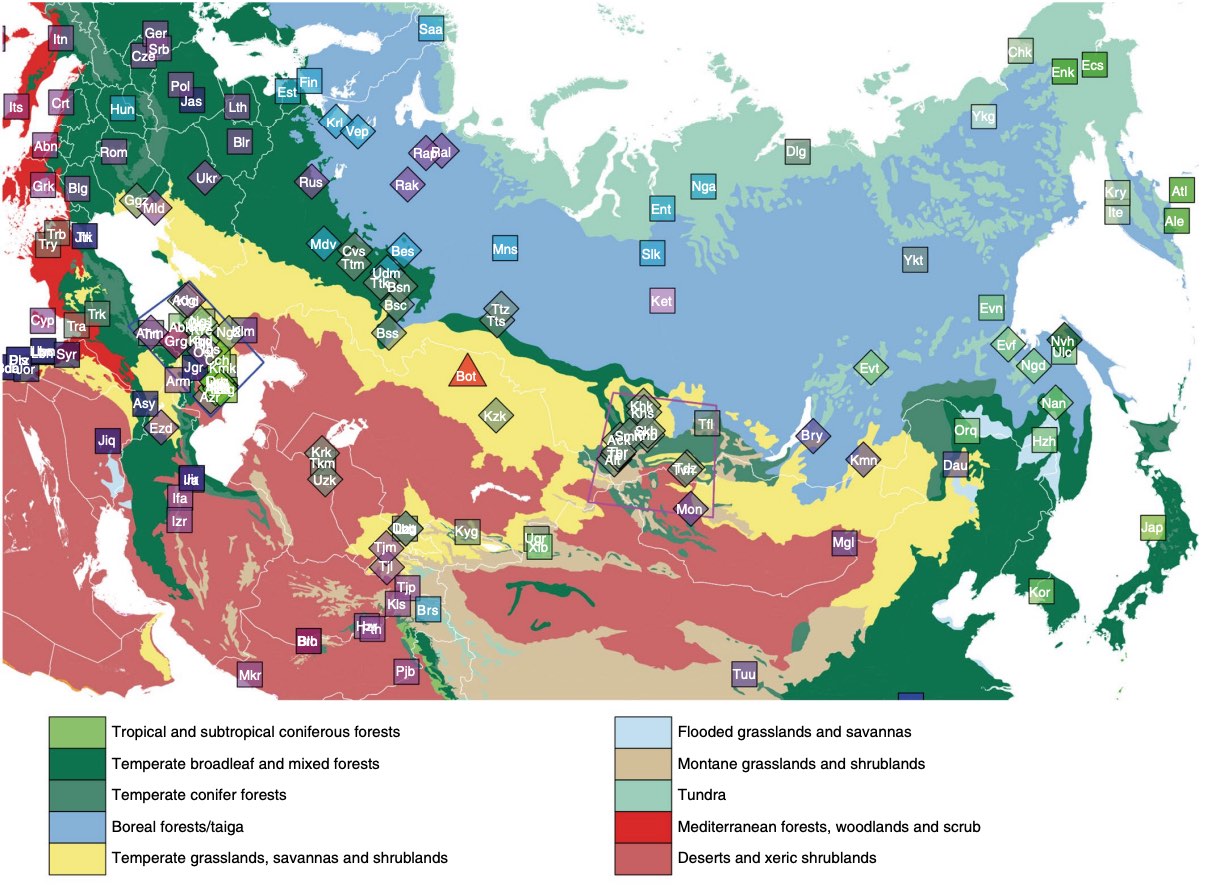
The indigenous populations of inner Eurasia-a huge geographic region covering the central Eurasian steppe and the northern Eurasian taiga and tundra-harbour tremendous diversity in their genes, cultures and languages. In this study, we report novel genome-wide data for 763 individuals from Armenia, Georgia, Kazakhstan, Moldova, Mongolia, Russia, Tajikistan, Ukraine and Uzbekistan. We furthermore report additional damage-reduced genome-wide data of two previously published individuals from the Eneolithic Botai culture in Kazakhstan (~5,400 BP). We find that present-day inner Eurasian populations are structured into three distinct admixture clines stretching between various western and eastern Eurasian ancestries, mirroring geography. The Botai and more recent ancient genomes from Siberia show a decrease in contributions from so-called 'ancient North Eurasian' ancestry over time, which is detectable only in the northern-most 'forest-tundra' cline. The intermediate 'steppe-forest' cline descends from the Late Bronze Age steppe ancestries, while the 'southern steppe' cline further to the south shows a strong West/South Asian influence. Ancient genomes suggest a northward spread of the southern steppe cline in Central Asia during the first millennium BC. Finally, the genetic structure of Caucasus populations highlights a role of the Caucasus Mountains as a barrier to gene flow and suggests a post-Neolithic gene flow into North Caucasus populations from the steppe.
Authors: Vanessa Villalba-Mouco, Marieke S van de Loosdrecht, Cosimo Posth, Rafael Mora, Jorge Martínez-Moreno, Manuel Rojo-Guerra, Domingo C Salazar-García, José I Royo-Guillén, Michael Kunst, Hélène Rougier, Isabelle Crevecoeur, Héctor Arcusa-Magallón, Cristina Tejedor-Rodríguez, Iñigo García-Martínez de Lagrán, Rafael Garrido-Pena, Kurt W Alt, Choongwon Jeong, Stephan Schiffels, Pilar Utrilla, Johannes Krause and Wolfgang Haak
Abstract:
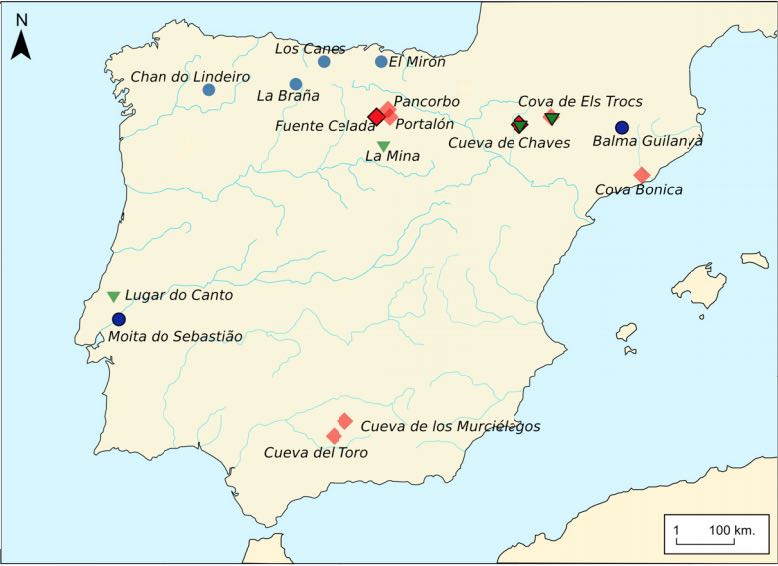
The Iberian Peninsula in southwestern Europe represents an important test case for the study of human population movements during prehistoric periods. During the Last Glacial Maximum (LGM), the peninsula formed a periglacial refugium [1] for hunter-gatherers (HGs) and thus served as a potential source for the re-peopling of northern latitudes [2]. The post-LGM genetic signature was previously described as a cline from Western HG (WHG) to Eastern HG (EHG), further shaped by later Holocene expansions from the Near East and the North Pontic steppes [3-9]. Western and central Europe were dominated by ancestry associated with the ∼14,000-year-old individual from Villabruna, Italy, which had largely replaced earlier genetic ancestry, represented by 19,000-15,000-year-old individuals associated with the Magdalenian culture [2]. However, little is known about the genetic diversity in southern European refugia, the presence of distinct genetic clusters, and correspondence with geography. Here, we report new genome-wide data from 11 HGs and Neolithic individuals that highlight the late survival of Paleolithic ancestry in Iberia, reported previously in Magdalenian-associated individuals. We show that all Iberian HGs, including the oldest, a ∼19,000-year-old individual from El Mirón in Spain, carry dual ancestry from both Villabruna and the Magdalenian-related individuals. Thus, our results suggest an early connection between two potential refugia, resulting in a genetic ancestry that survived in later Iberian HGs. Our new genomic data from Iberian Early and Middle Neolithic individuals show that the dual Iberian HG genomic legacy pertains in the peninsula, suggesting that expanding farmers mixed with local HGs. VIDEO ABSTRACT.
Authors: Chuan-Chao Wang, Sabine Reinhold, Alexey Kalmykov, Antje Wissgott, Guido Brandt, Choongwon Jeong, Olivia Cheronet, Matthew Ferry, Eadaoin Harney, Denise Keating, Swapan Mallick, Nadin Rohland, Kristin Stewardson, Anatoly R Kantorovich, Vladimir E Maslov, Vladimira G Petrenko, Vladimir R Erlikh, Biaslan Ch Atabiev, Rabadan G Magomedov, Philipp L Kohl, Kurt W Alt, Sandra L Pichler, Claudia Gerling, Harald Meller, Benik Vardanyan, Larisa Yeganyan, Alexey D Rezepkin, Dirk Mariaschk, Natalia Berezina, Julia Gresky, Katharina Fuchs, Corina Knipper, Stephan Schiffels, Elena Balanovska, Oleg Balanovsky, Iain Mathieson, Thomas Higham, Yakov B Berezin, Alexandra Buzhilova, Viktor Trifonov, Ron Pinhasi, Andrej B Belinskij, David Reich, Svend Hansen, Johannes Krause and Wolfgang Haak
Abstract:
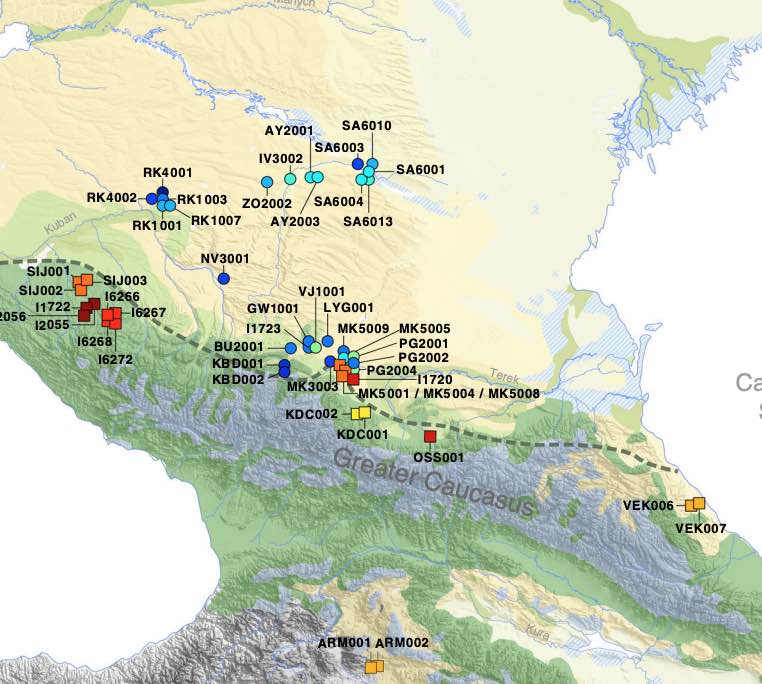
Archaeogenetic studies have described the formation of Eurasian 'steppe ancestry' as a mixture of Eastern and Caucasus hunter-gatherers. However, it remains unclear when and where this ancestry arose and whether it was related to a horizon of cultural innovations in the 4th millennium BCE that subsequently facilitated the advance of pastoral societies in Eurasia. Here we generated genome-wide SNP data from 45 prehistoric individuals along a 3000-year temporal transect in the North Caucasus. We observe a genetic separation between the groups of the Caucasus and those of the adjacent steppe. The northern Caucasus groups are genetically similar to contemporaneous populations south of it, suggesting human movement across the mountain range during the Bronze Age. The steppe groups from Yamnaya and subsequent pastoralist cultures show evidence for previously undetected farmer-related ancestry from different contact zones, while Steppe Maykop individuals harbour additional Upper Palaeolithic Siberian and Native American related ancestry.
Authors: Thiseas C Lamnidis, Kerttu Majander, Choongwon Jeong, Elina Salmela, Anna Wessman, Vyacheslav Moiseyev, Valery Khartanovich, Oleg Balanovsky, Matthias Ongyerth, Antje Weihmann, Antti Sajantila, Janet Kelso, Svante Pääbo, Päivi Onkamo, Wolfgang Haak, Johannes Krause and Stephan Schiffels
Abstract:
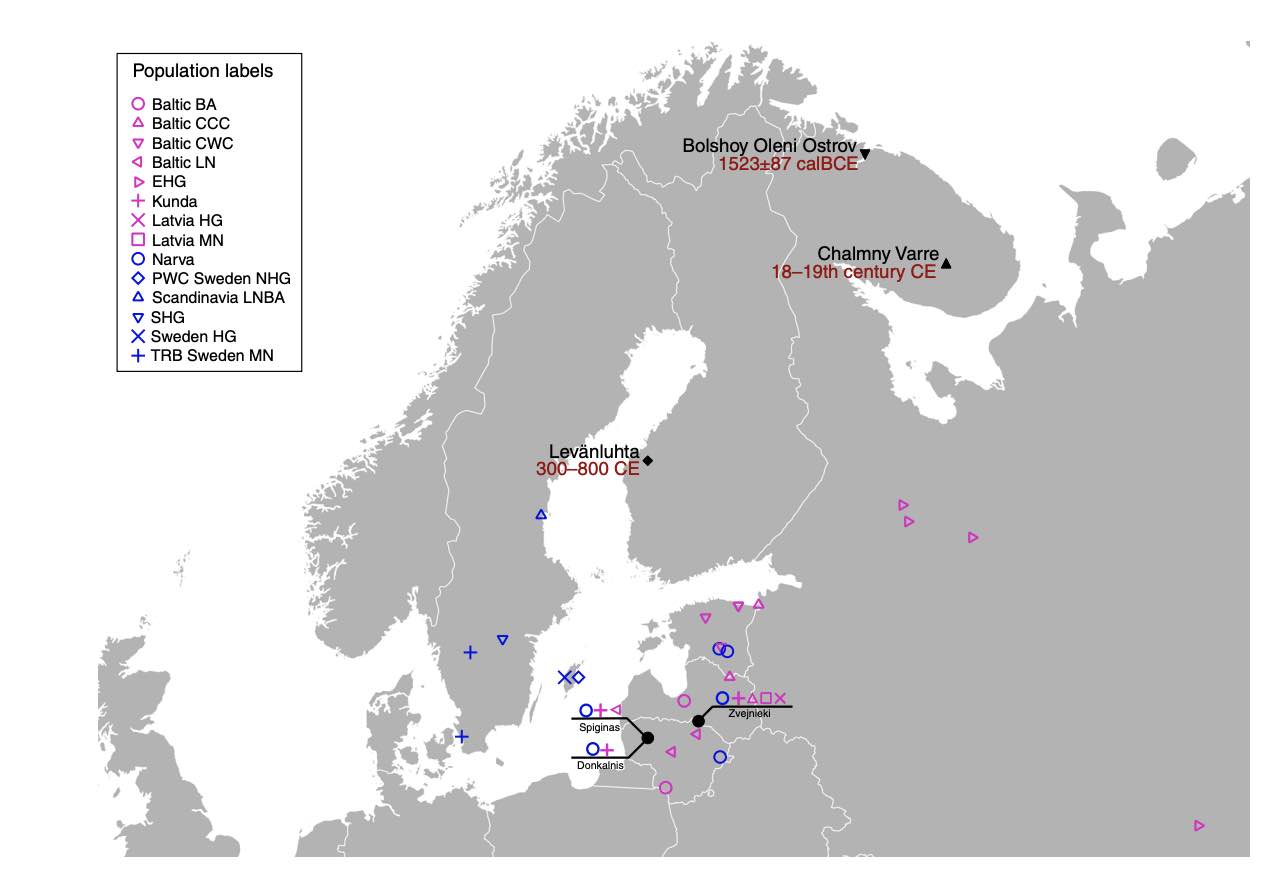
European population history has been shaped by migrations of people, and their subsequent admixture. Recently, ancient DNA has brought new insights into European migration events linked to the advent of agriculture, and possibly to the spread of Indo-European languages. However, little is known about the ancient population history of north-eastern Europe, in particular about populations speaking Uralic languages, such as Finns and Saami. Here we analyse ancient genomic data from 11 individuals from Finland and north-western Russia. We show that the genetic makeup of northern Europe was shaped by migrations from Siberia that began at least 3500 years ago. This Siberian ancestry was subsequently admixed into many modern populations in the region, particularly into populations speaking Uralic languages today. Additionally, we show that ancestors of modern Saami inhabited a larger territory during the Iron Age, which adds to the historical and linguistic information about the population history of Finland.
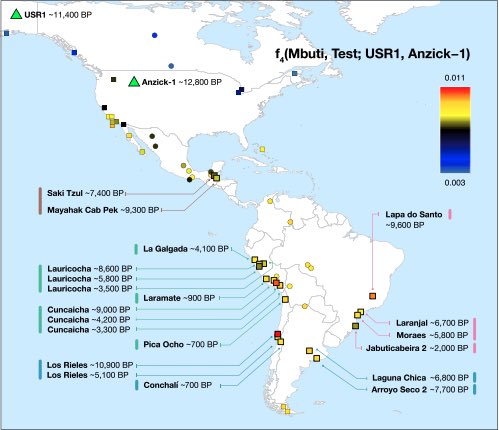
Authors: Cosimo Posth, Nathan Nakatsuka, Iosif Lazaridis, Pontus Skoglund, Swapan Mallick, Thiseas C Lamnidis, Nadin Rohland, Kathrin Nägele, Nicole Adamski, Emilie Bertolini, Nasreen Broomandkhoshbacht, Alan Cooper, Brendan J Culleton, Tiago Ferraz, Matthew Ferry, Anja Furtwängler, Wolfgang Haak, Kelly Harkins, Thomas K Harper, Tábita Hünemeier, Ann Marie Lawson, Bastien Llamas, Megan Michel, Elizabeth Nelson, Jonas Oppenheimer, Nick Patterson, Stephan Schiffels, Jakob Sedig, Kristin Stewardson, Sahra Talamo, Chuan-Chao Wang, Jean-Jacques Hublin, Mark Hubbe, Katerina Harvati, Amalia Nuevo Delaunay, Judith Beier, Michael Francken, Peter Kaulicke, Hugo Reyes-Centeno, Kurt Rademaker, Willa R Trask, Mark Robinson, Said M Gutierrez, Keith M Prufer, Domingo C Salazar-García, Eliane N Chim, Lisiane Müller Plumm Gomes, Marcony L Alves, Andersen Liryo, Mariana Inglez, Rodrigo E Oliveira, Danilo V Bernardo, Alberto Barioni, Veronica Wesolowski, Nahuel A Scheifler, Mario A Rivera, Claudia R Plens, Pablo G Messineo, Levy Figuti, Daniel Corach, Clara Scabuzzo, Sabine Eggers, Paulo DeBlasis, Markus Reindel, César Méndez, Gustavo Politis, Elsa Tomasto-Cagigao, Douglas J Kennett, André Strauss, Lars Fehren-Schmitz, Johannes Krause and David Reich
Abstract:
We report genome-wide ancient DNA from 49 individuals forming four parallel time transects in Belize, Brazil, the Central Andes, and the Southern Cone, each dating to at least ∼9,000 years ago. The common ancestral population radiated rapidly from just one of the two early branches that contributed to Native Americans today. We document two previously unappreciated streams of gene flow between North and South America. One affected the Central Andes by ∼4,200 years ago, while the other explains an affinity between the oldest North American genome associated with the Clovis culture and the oldest Central and South Americans from Chile, Brazil, and Belize. However, this was not the primary source for later South Americans, as the other ancient individuals derive from lineages without specific affinity to the Clovis-associated genome, suggesting a population replacement that began at least 9,000 years ago and was followed by substantial population continuity in multiple regions.
Authors: Ben A Potter, James F Baichtal, Alwynne B Beaudoin, Lars Fehren-Schmitz, C Vance Haynes, Vance T Holliday, Charles E Holmes, John W Ives, Robert L Kelly, Bastien Llamas, Ripan S Malhi, D Shane Miller, David Reich, Joshua D Reuther, Stephan Schiffels and Todd A Surovell
Authors: Ben A Potter, Alwynne B Beaudoin, C Vance Haynes, Vance T Holliday, Charles E Holmes, John W Ives, Robert Kelly, Bastien Llamas, Ripan Malhi, Shane Miller, David Reich, Joshua D Reuther, Stephan Schiffels and Todd Surovell
Authors: Marieke van de Loosdrecht, Abdeljalil Bouzouggar, Louise Humphrey, Cosimo Posth, Nick Barton, Ayinuer Aximu-Petri, Birgit Nickel, Sarah Nagel, El Hassan Talbi, Mohammed Abdeljalil El Hajraoui, Saaïd Amzazi, Jean-Jacques Hublin, Svante Pääbo, Stephan Schiffels, Matthias Meyer, Wolfgang Haak, Choongwon Jeong and Johannes Krause
Abstract:
North Africa is a key region for understanding human history, but the genetic history of its people is largely unknown. We present genomic data from seven 15,000-year-old modern humans from Morocco, attributed to the Iberomaurusian culture. We find a genetic affinity with early Holocene Near Easterners, best represented by Levantine Natufians, suggesting a pre-agricultural connection between Africa and the Near East. We do not find evidence for gene flow from Paleolithic Europeans into Late Pleistocene North Africans. The Taforalt individuals derive one third of their ancestry from sub-Saharan Africans, best approximated by a mixture of genetic components preserved in present-day West and East Africans. Thus, we provide direct evidence for genetic interactions between modern humans across Africa and Eurasia in the Pleistocene.
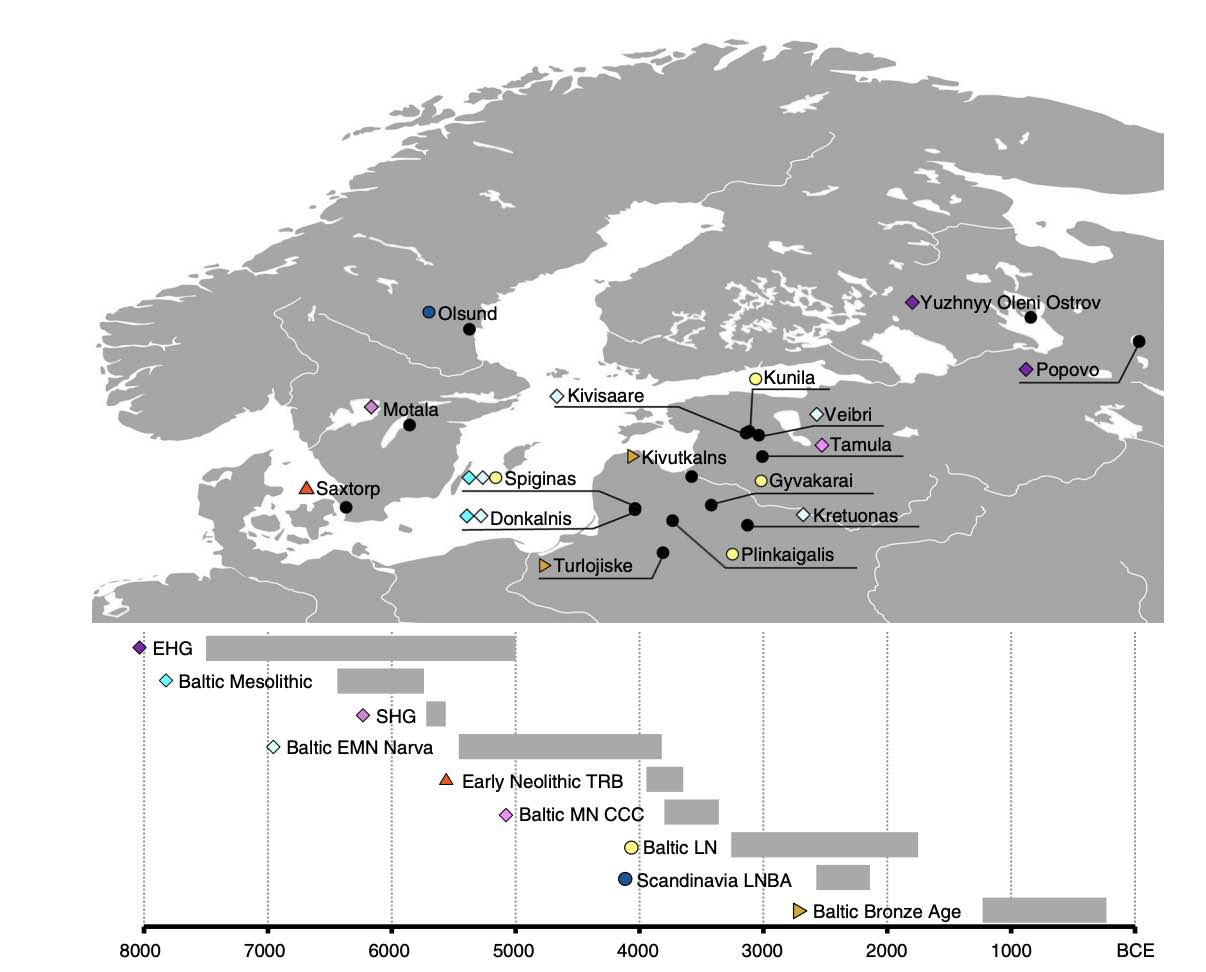
Authors: Alissa Mittnik, Chuan-Chao Wang, Saskia Pfrengle, Mantas Daubaras, Gunita Zariņa, Fredrik Hallgren, Raili Allmäe, Valery Khartanovich, Vyacheslav Moiseyev, Mari Tõrv, Anja Furtwängler, Aida Andrades Valtueña, Michal Feldman, Christos Economou, Markku Oinonen, Andrejs Vasks, Elena Balanovska, David Reich, Rimantas Jankauskas, Wolfgang Haak, Stephan Schiffels and Johannes Krause
Abstract:
While the series of events that shaped the transition between foraging societies and food producers are well described for Central and Southern Europe, genetic evidence from Northern Europe surrounding the Baltic Sea is still sparse. Here, we report genome-wide DNA data from 38 ancient North Europeans ranging from ~9500 to 2200 years before present. Our analysis provides genetic evidence that hunter-gatherers settled Scandinavia via two routes. We reveal that the first Scandinavian farmers derive their ancestry from Anatolia 1000 years earlier than previously demonstrated. The range of Mesolithic Western hunter-gatherers extended to the east of the Baltic Sea, where these populations persisted without gene-flow from Central European farmers during the Early and Middle Neolithic. The arrival of steppe pastoralists in the Late Neolithic introduced a major shift in economy and mediated the spread of a new ancestry associated with the Corded Ware Complex in Northern Europe.
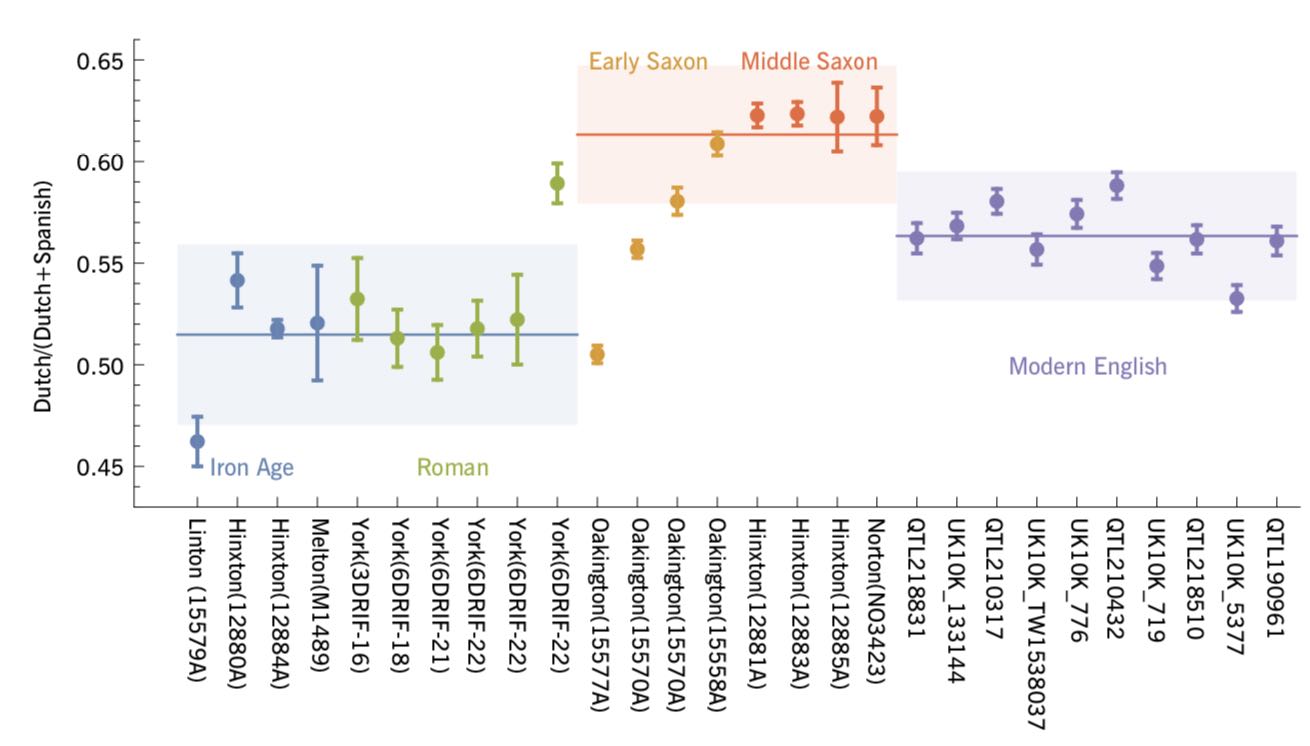
Authors: Stephan Schiffels and Duncan Sayer
Abstract:
British population history has been shaped by a series of immigration periods, including the Roman occupation from AD 43 and Early Anglo-Saxon migrations after AD 410. Until recently, it was an open question to whatthe extent to which these events changed the genetic population structure in Britain was an open question. Two recent genetic studies have provided new insights into this question, by analysing the genome sequences from 19 ancient British samples in total, dating from between the 1st century BC until and the 8th century AD . Here we will review these two recent studies, present a joint analysis of all 19 available samples, and will put the results into a wider archaeological context. Key results reviewed here include: 1 High high levels of genetic continuity between the late Late Iron Age and the Romano-British period; 2 a clear increase in ancestry related to the modern Dutch population during the Anglo-Saxon period, suggesting a substantial arrival of new people during this time; 3 an estimated 38 \% average of the proportion of Anglo-Saxon ancestry in the modern English population at 38\% on average; 4 evidence from an Early Anglo-Saxon cemetery near Cambridge for of early admixture between of Anglo-Saxon immigrants and indigenous British inhabitants from an early Anglo-Saxon cemetery near Cambridge.
Authors: Verena J Schuenemann, Alexander Peltzer, Beatrix Welte, W Paul van Pelt, Martyna Molak, Chuan-Chao Wang, Anja Furtwängler, Christian Urban, Ella Reiter, Kay Nieselt, Barbara Teßmann, Michael Francken, Katerina Harvati, Wolfgang Haak, Stephan Schiffels and Johannes Krause
Abstract:
Egypt, located on the isthmus of Africa, is an ideal region to study historical population dynamics due to its geographic location and documented interactions with ancient civilizations in Africa, Asia and Europe. Particularly, in the first millennium BCE Egypt endured foreign domination leading to growing numbers of foreigners living within its borders possibly contributing genetically to the local population. Here we present 90 mitochondrial genomes as well as genome-wide data sets from three individuals obtained from Egyptian mummies. The samples recovered from Middle Egypt span around 1,300 years of ancient Egyptian history from the New Kingdom to the Roman Period. Our analyses reveal that ancient Egyptians shared more ancestry with Near Easterners than present-day Egyptians, who received additional sub-Saharan admixture in more recent times. This analysis establishes ancient Egyptian mummies as a genetic source to study ancient human history and offers the perspective of deciphering Egypt's past at a genome-wide level.
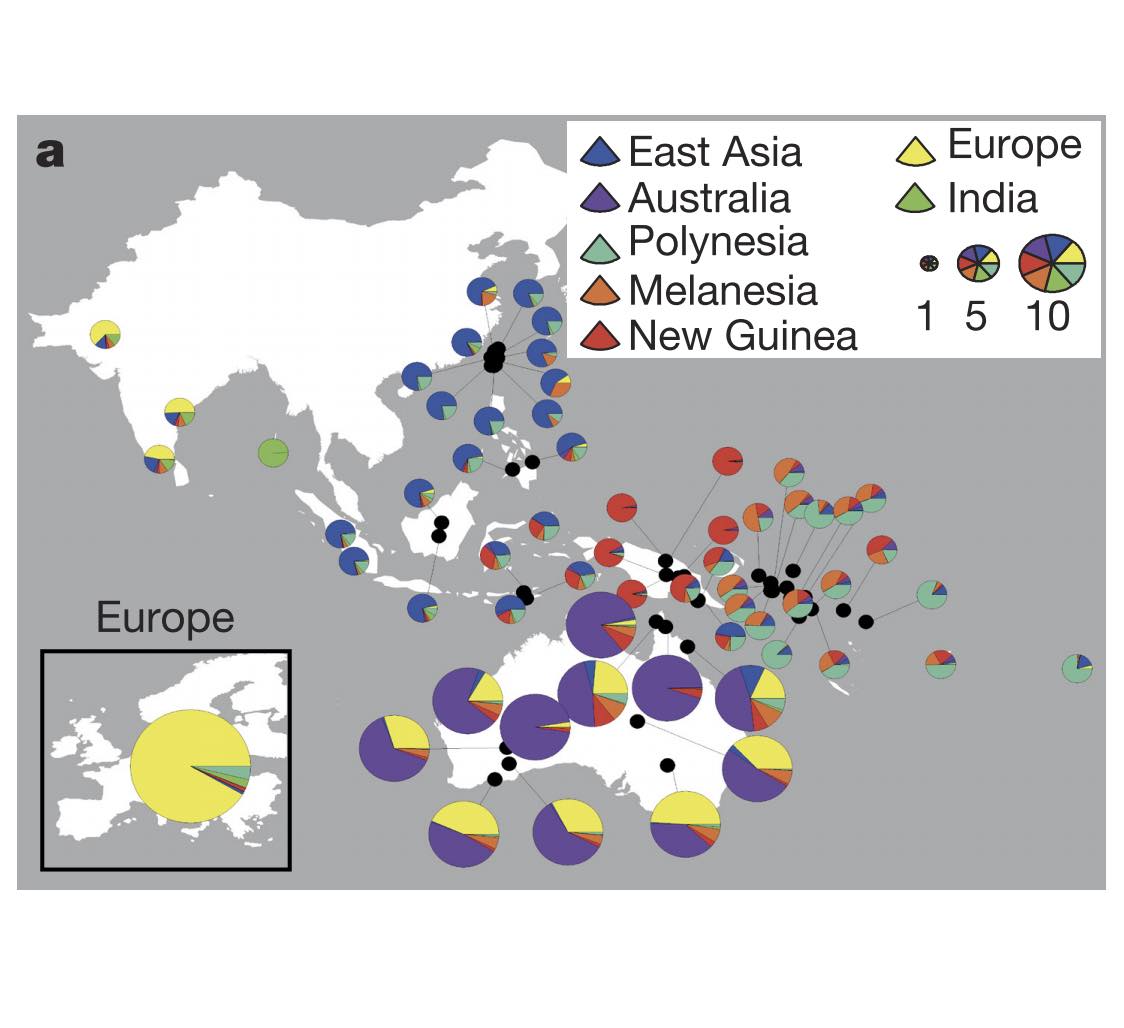
Authors: Anna-Sapfo Malaspinas, Michael C Westaway, Craig Muller, Vitor C Sousa, Oscar Lao, Isabel Alves, Anders Bergström, Georgios Athanasiadis, Jade Y Cheng, Jacob E Crawford, Tim H Heupink, Enrico Macholdt, Stephan Peischl, Simon Rasmussen, Stephan Schiffels, Sankar Subramanian, Joanne L Wright, Anders Albrechtsen, Chiara Barbieri, Isabelle Dupanloup, Anders Eriksson, Ashot Margaryan, Ida Moltke, Irina Pugach, Thorfinn S Korneliussen, Ivan P Levkivskyi, J Víctor Moreno-Mayar, Shengyu Ni, Fernando Racimo, Martin Sikora, Yali Xue, Farhang A Aghakhanian, Nicolas Brucato, Søren Brunak, Paula F Campos, Warren Clark, Sturla Ellingvåg, Gudjugudju Fourmile, Pascale Gerbault, Darren Injie, George Koki, Matthew Leavesley, Betty Logan, Aubrey Lynch, Elizabeth A Matisoo-Smith, Peter J McAllister, Alexander J Mentzer, Mait Metspalu, Andrea B Migliano, Les Murgha, Maude E Phipps, William Pomat, Doc Reynolds, François-Xavier Ricaut, Peter Siba, Mark G Thomas, Thomas Wales, Colleen Ma'run Wall, Stephen J Oppenheimer, Chris Tyler-Smith, Richard Durbin, Joe Dortch, Andrea Manica, Mikkel H Schierup, Robert A Foley, Marta Mirazon Lahr, Claire Bowern, Jeffrey D Wall, Thomas Mailund, Mark Stoneking, Rasmus Nielsen, Manjinder S Sandhu, Laurent Excoffier, David M Lambert and Eske Willerslev
Abstract:
The population history of Aboriginal Australians remains largely uncharacterized. Here we generate high-coverage genomes for 83 Aboriginal Australians (speakers of Pama-Nyungan languages) and 25 Papuans from the New Guinea Highlands. We find that Papuan and Aboriginal Australian ancestors diversified 25-40 thousand years ago (kya), suggesting pre-Holocene population structure in the ancient continent of Sahul (Australia, New Guinea and Tasmania). However, all of the studied Aboriginal Australians descend from a single founding population that differentiated ~10-32 kya. We infer a population expansion in northeast Australia during the Holocene epoch (past 10,000 years) associated with limited gene flow from this region to the rest of Australia, consistent with the spread of the Pama-Nyungan languages. We estimate that Aboriginal Australians and Papuans diverged from Eurasians 51-72 kya, following a single out-of-Africa dispersal, and subsequently admixed with archaic populations. Finally, we report evidence of selection in Aboriginal Australians potentially associated with living in the desert.
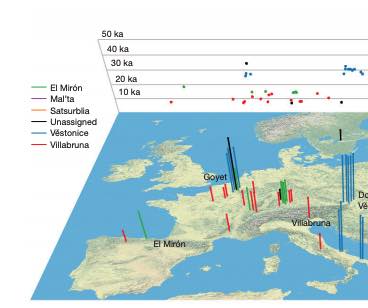
Authors: Qiaomei Fu, Cosimo Posth, Mateja Hajdinjak, Martin Petr, Swapan Mallick, Daniel Fernandes, Anja Furtwängler, Wolfgang Haak, Matthias Meyer, Alissa Mittnik, Birgit Nickel, Alexander Peltzer, Nadin Rohland, Viviane Slon, Sahra Talamo, Iosif Lazaridis, Mark Lipson, Iain Mathieson, Stephan Schiffels, Pontus Skoglund, Anatoly P Derevianko, Nikolai Drozdov, Vyacheslav Slavinsky, Alexander Tsybankov, Renata Grifoni Cremonesi, Francesco Mallegni, Bernard Gély, Eligio Vacca, Manuel R González Morales, Lawrence G Straus, Christine Neugebauer-Maresch, Maria Teschler-Nicola, Silviu Constantin, Oana Teodora Moldovan, Stefano Benazzi, Marco Peresani, Donato Coppola, Martina Lari, Stefano Ricci, Annamaria Ronchitelli, Frédérique Valentin, Corinne Thevenet, Kurt Wehrberger, Dan Grigorescu, Hélène Rougier, Isabelle Crevecoeur, Damien Flas, Patrick Semal, Marcello A Mannino, Christophe Cupillard, Hervé Bocherens, Nicholas J Conard, Katerina Harvati, Vyacheslav Moiseyev, Dorothée G Drucker, Jiří Svoboda, Michael P Richards, David Caramelli, Ron Pinhasi, Janet Kelso, Nick Patterson, Johannes Krause, Svante Pääbo and David E Reich
Abstract:
Modern humans arrived in Europe ~45,000 years ago, but little is known about their genetic composition before the start of farming ~8,500 years ago. Here we analyse genome-wide data from 51 Eurasians from ~45,000-7,000 years ago. Over this time, the proportion of Neanderthal DNA decreased from 3-6\% to around 2\%, consistent with natural selection against Neanderthal variants in modern humans. Whereas there is no evidence of the earliest modern humans in Europe contributing to the genetic composition of present-day Europeans, all individuals between ~37,000 and ~14,000 years ago descended from a single founder population which forms part of the ancestry of present-day Europeans. An ~35,000-year-old individual from northwest Europe represents an early branch of this founder population which was then displaced across a broad region, before reappearing in southwest Europe at the height of the last Ice Age ~19,000 years ago. During the major warming period after ~14,000 years ago, a genetic component related to present-day Near Easterners became widespread in Europe. These results document how population turnover and migration have been recurring themes of European prehistory.
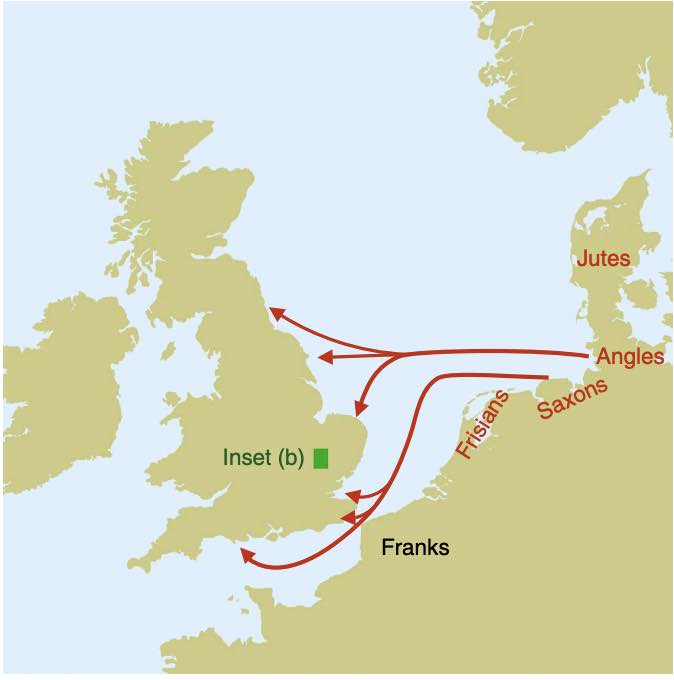
Authors: Stephan Schiffels, Wolfgang Haak, Pirita Paajanen, Bastien Llamas, Elizabeth Popescu, Louise Loe, Rachel Clarke, Alice Lyons, Richard Mortimer, Duncan Sayer, Chris Tyler-Smith, Alan Cooper and Richard Durbin
Abstract:
British population history has been shaped by a series of immigrations, including the early Anglo-Saxon migrations after 400 CE. It remains an open question how these events affected the genetic composition of the current British population. Here, we present whole-genome sequences from 10 individuals excavated close to Cambridge in the East of England, ranging from the late Iron Age to the middle Anglo-Saxon period. By analysing shared rare variants with hundreds of modern samples from Britain and Europe, we estimate that on average the contemporary East English population derives 38\% of its ancestry from Anglo-Saxon migrations. We gain further insight with a new method, rarecoal, which infers population history and identifies fine-scale genetic ancestry from rare variants. Using rarecoal we find that the Anglo-Saxon samples are closely related to modern Dutch and Danish populations, while the Iron Age samples share ancestors with multiple Northern European populations including Britain.
Authors: Milan Malinsky, Richard J Challis, Alexandra M Tyers, Stephan Schiffels, Yohey Terai, Benjamin P Ngatunga, Eric A Miska, Richard Durbin, Martin J Genner and George F Turner
Abstract:
The genomic causes and effects of divergent ecological selection during speciation are still poorly understood. Here we report the discovery and detailed characterization of early-stage adaptive divergence of two cichlid fish ecomorphs in a small (700 meters in diameter) isolated crater lake in Tanzania. The ecomorphs differ in depth preference, male breeding color, body shape, diet, and trophic morphology. With whole-genome sequences of 146 fish, we identified 98 clearly demarcated genomic ``islands'' of high differentiation and demonstrated the association of genotypes across these islands with divergent mate preferences. The islands contain candidate adaptive genes enriched for functions in sensory perception (including rhodopsin and other twilight-vision-associated genes), hormone signaling, and morphogenesis. Our study suggests mechanisms and genomic regions that may play a role in the closely related mega-radiation of Lake Malawi.
Authors: UK10K Consortium , Klaudia Walter, Josine L Min, Jie Huang, Lucy Crooks, Yasin Memari, Shane McCarthy, John R B Perry, Changjiang Xu, Marta Futema, Daniel Lawson, Valentina Iotchkova, Stephan Schiffels, Audrey E Hendricks, Petr Danecek, Rui Li, James Floyd, Louise V Wain, Inês Barroso, Steve E Humphries, Matthew E Hurles, Eleftheria Zeggini, Jeffrey C Barrett, Vincent Plagnol, J Brent Richards, Celia M T Greenwood, Nicholas J Timpson, Richard Durbin and Nicole Soranzo
Abstract:
The contribution of rare and low-frequency variants to human traits is largely unexplored. Here we describe insights from sequencing whole genomes (low read depth, 7×) or exomes (high read depth, 80×) of nearly 10,000 individuals from population-based and disease collections. In extensively phenotyped cohorts we characterize over 24 million novel sequence variants, generate a highly accurate imputation reference panel and identify novel alleles associated with levels of triglycerides (APOB), adiponectin (ADIPOQ) and low-density lipoprotein cholesterol (LDLR and RGAG1) from single-marker and rare variant aggregation tests. We describe population structure and functional annotation of rare and low-frequency variants, use the data to estimate the benefits of sequencing for association studies, and summarize lessons from disease-specific collections. Finally, we make available an extensive resource, including individual-level genetic and phenotypic data and web-based tools to facilitate the exploration of association results.
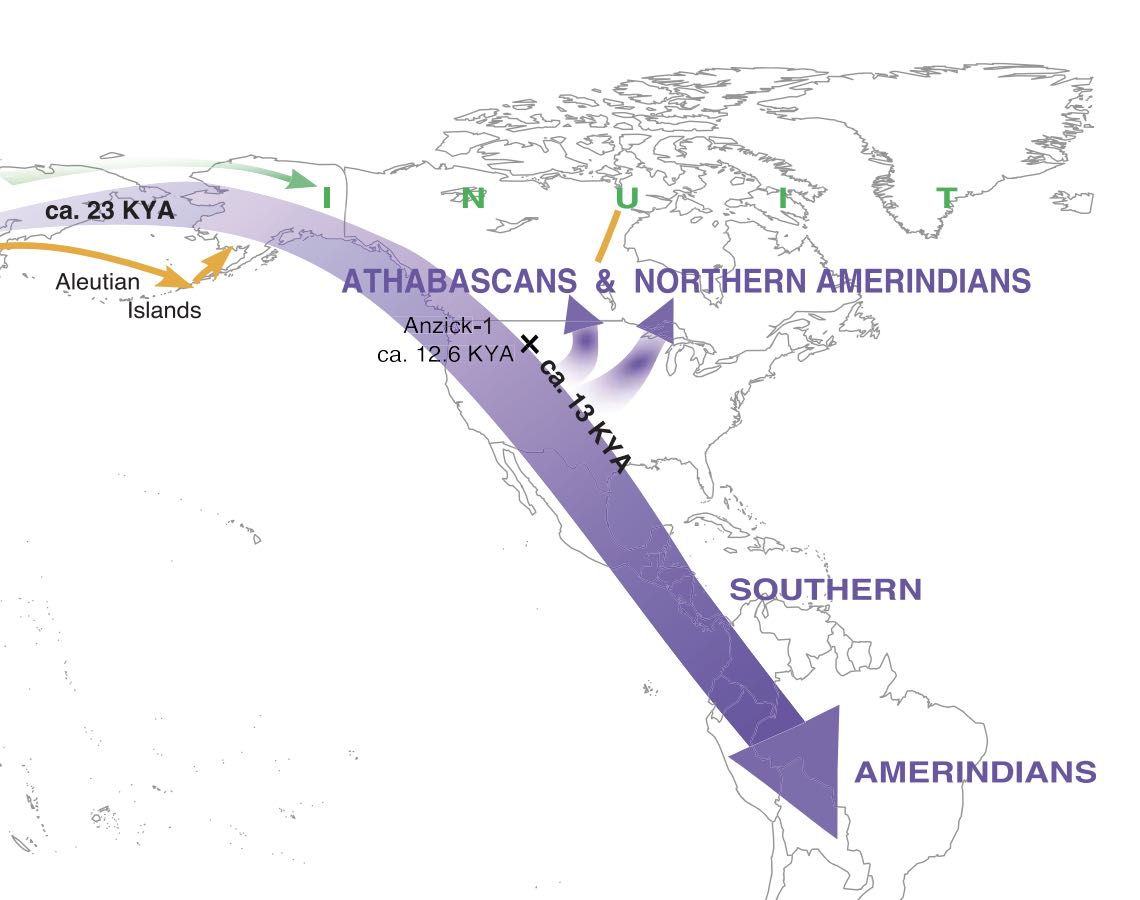
Authors: Maanasa Raghavan, Matthias Steinrücken, Kelley Harris, Stephan Schiffels, Simon Rasmussen, Michael DeGiorgio, Anders Albrechtsen, Cristina Valdiosera, María C Ávila-Arcos, Anna-Sapfo Malaspinas, Anders Eriksson, Ida Moltke, Mait Metspalu, Julian R Homburger, Jeff Wall, Omar E Cornejo, J Víctor Moreno-Mayar, Thorfinn S Korneliussen, Tracey Pierre, Morten Rasmussen, Paula F Campos, Peter de Barros Damgaard, Morten E Allentoft, John Lindo, Ene Metspalu, Ricardo Rodríguez-Varela, Josefina Mansilla, Celeste Henrickson, Andaine Seguin-Orlando, Helena Malmström, Thomas Stafford, Suyash S Shringarpure, Andres Moreno-Estrada, Monika Karmin, Kristiina Tambets, Anders Bergström, Yali Xue, Vera Warmuth, Andrew D Friend, Joy Singarayer, Paul Valdes, Francois Balloux, Ilán Leboreiro, Jose Luis Vera, Hector Rangel-Villalobos, Davide Pettener, Donata Luiselli, Loren G Davis, Evelyne Heyer, Christoph P E Zollikofer, Marcia S Ponce de León, Colin I Smith, Vaughan Grimes, Kelly-Anne Pike, Michael Deal, Benjamin T Fuller, Bernardo Arriaza, Vivien Standen, Maria F Luz, Francois Ricaut, Niede Guidon, Ludmila Osipova, Mikhail I Voevoda, Olga L Posukh, Oleg Balanovsky, Maria Lavryashina, Yuri Bogunov, Elza Khusnutdinova, Marina Gubina, Elena Balanovska, Sardana Fedorova, Sergey Litvinov, Boris Malyarchuk, Miroslava Derenko, M J Mosher, David Archer, Jerome Cybulski, Barbara Petzelt, Joycelynn Mitchell, Rosita Worl, Paul J Norman, Peter Parham, Brian M Kemp, Toomas Kivisild, Chris Tyler-Smith, Manjinder S Sandhu, Michael Crawford, Richard Villems, David Glenn Smith, Michael R Waters, Ted Goebel, John R Johnson, Ripan S Malhi, Mattias Jakobsson, David J Meltzer, Andrea Manica, Richard Durbin, Carlos D Bustamante, Yun S Song, Rasmus Nielsen and Eske Willerslev
Abstract:
How and when the Americas were populated remains contentious. Using ancient and modern genome-wide data, we found that the ancestors of all present-day Native Americans, including Athabascans and Amerindians, entered the Americas as a single migration wave from Siberia no earlier than 23 thousand years ago (ka) and after no more than an 8000-year isolation period in Beringia. After their arrival to the Americas, ancestral Native Americans diversified into two basal genetic branches around 13 ka, one that is now dispersed across North and South America and the other restricted to North America. Subsequent gene flow resulted in some Native Americans sharing ancestry with present-day East Asians (including Siberians) and, more distantly, Australo-Melanesians. Putative ``Paleoamerican'' relict populations, including the historical Mexican Pericúes and South American Fuego-Patagonians, are not directly related to modern Australo-Melanesians as suggested by the Paleoamerican Model.
Authors: Luca Pagani, Stephan Schiffels, Deepti Gurdasani, Petr Danecek, Aylwyn Scally, Yuan Chen, Yali Xue, Marc Haber, Rosemary Ekong, Tamiru Oljira, Ephrem Mekonnen, Donata Luiselli, Neil Bradman, Endashaw Bekele, Pierre Zalloua, Richard Durbin, Toomas Kivisild and Chris Tyler-Smith
Abstract:
The predominantly African origin of all modern human populations is well established, but the route taken out of Africa is still unclear. Two alternative routes, via Egypt and Sinai or across the Bab el Mandeb strait into Arabia, have traditionally been proposed as feasible gateways in light of geographic, paleoclimatic, archaeological, and genetic evidence. Distinguishing among these alternatives has been difficult. We generated 225 whole-genome sequences (225 at 8× depth, of which 8 were increased to 30×; Illumina HiSeq 2000) from six modern Northeast African populations (100 Egyptians and five Ethiopian populations each represented by 25 individuals). West Eurasian components were masked out, and the remaining African haplotypes were compared with a panel of sub-Saharan African and non-African genomes. We showed that masked Northeast African haplotypes overall were more similar to non-African haplotypes and more frequently present outside Africa than were any sets of haplotypes derived from a West African population. Furthermore, the masked Egyptian haplotypes showed these properties more markedly than the masked Ethiopian haplotypes, pointing to Egypt as the more likely gateway in the exodus to the rest of the world. Using five Ethiopian and three Egyptian high-coverage masked genomes and the multiple sequentially Markovian coalescent (MSMC) approach, we estimated the genetic split times of Egyptians and Ethiopians from non-African populations at 55,000 and 65,000 years ago, respectively, whereas that of West Africans was estimated to be 75,000 years ago. Both the haplotype and MSMC analyses thus suggest a predominant northern route out of Africa via Egypt.
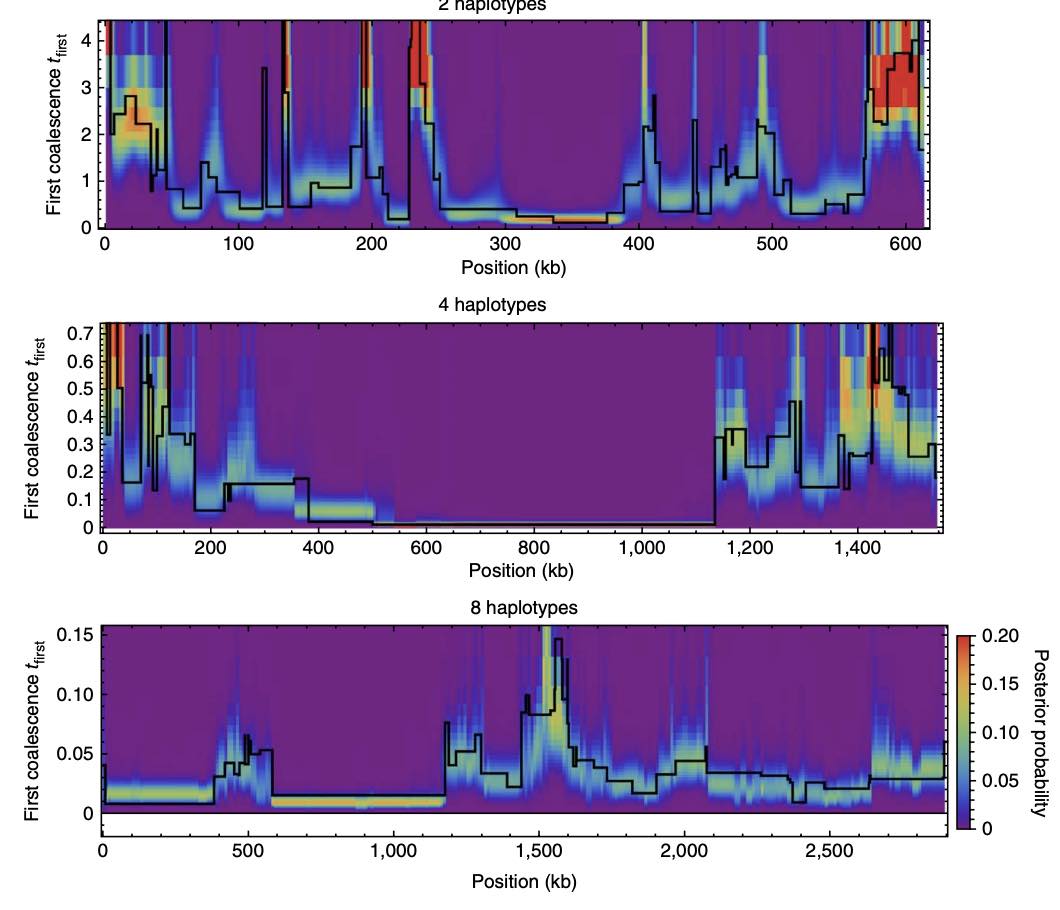
Authors: Stephan Schiffels and Richard Durbin
Abstract:
The availability of complete human genome sequences from populations across the world has given rise to new population genetic inference methods that explicitly model ancestral relationships under recombination and mutation. So far, application of these methods to evolutionary history more recent than 20,000-30,000 years ago and to population separations has been limited. Here we present a new method that overcomes these shortcomings. The multiple sequentially Markovian coalescent (MSMC) analyzes the observed pattern of mutations in multiple individuals, focusing on the first coalescence between any two individuals. Results from applying MSMC to genome sequences from nine populations across the world suggest that the genetic separation of non-African ancestors from African Yoruban ancestors started long before 50,000 years ago and give information about human population history as recent as 2,000 years ago, including the bottleneck in the peopling of the Americas and separations within Africa, East Asia and Europe.
Authors: Armita Nourmohammad, Stephan Schiffels and Michael Lässig
Abstract:
Molecular phenotypes are important links between genomic information and organismic functions, fitness, and evolution. Complex phenotypes, which are also called quantitative traits, often depend on multiple genomic loci. Their evolution builds on genome evolution in a complicated way, which involves selection, genetic drift, mutations and recombination. Here we develop a coarse-grained evolutionary statistics for phenotypes, which decouples from details of the underlying genotypes. We derive approximate evolution equations for the distribution of phenotype values within and across populations. This dynamics covers evolutionary processes at high and low recombination rates, that is, it applies to sexual and asexual populations. In a fitness landscape with a single optimal phenotype value, the phenotypic diversity within populations and the divergence between populations reach evolutionary equilibria, which describe stabilizing selection. We compute the equilibrium distributions of both quantities analytically and we show that the ratio of mean divergence and diversity depends on the strength of selection in a universal way: it is largely independent of the phenotype's genomic encoding and of the recombination rate. This establishes a new method for the inference of selection on molecular phenotypes beyond the genome level. We discuss the implications of our findings for the predictability of evolutionary processes.
Authors: Christopher J R Illingworth, Leopold Parts, Stephan Schiffels, Gianni Liti and Ville Mustonen
Abstract:
When selection is acting on a large, genetically diverse population, beneficial alleles increase in frequency. This fact can be used to map quantitative trait loci by sequencing the pooled DNA from the population at consecutive time points, and observing allele frequency changes. Here we present a population genetic method to analyse time-series data of allele frequencies from such an experiment. Beginning with a range of proposed evolutionary scenarios, the method measures the consistency of each with the observed frequency changes. Evolutionary theory is utilized to formulate equations of motion for the allele frequencies, following which likelihoods for having observed the sequencing data under each scenario are derived. Comparison of these likelihoods gives an insight into the prevailing dynamics of the system under study. We illustrate the method by quantifying selective effects from an experiment in which two phenotypically different yeast strains were first crossed and then propagated under heat stress (Parts et al., Genome Res. 2011). From these data we discover that about 6\% of polymorphic sites evolve non-neutrally under heat stress condition, either because of their linkage to beneficial (driver) alleles or because they are drivers themselves. We further identify 44 genomic regions containing one or more candidate driver alleles, quantify their apparent selective advantage, obtain estimates of recombination rates within the regions, and show that the dynamics of the drivers display a strong signature of selection going beyond additive models. Our approach is applicable to study adaptation in a range of systems under different evolutionary pressures.
Authors: Stephan Schiffels, Gergely Szöllösi, Ville Mustonen and Michael Lässig
Abstract:
In non-recombining genomes, genetic linkage can be an important evolutionary force. Linkage generates interference interactions, by which simultaneously occurring mutations affect each other's chance of fixation. Here, we develop a comprehensive model of adaptive evolution in linked genomes, which integrates interference interactions between multiple beneficial and deleterious mutations into a unified framework. By an approximate analytical solution, we predict the fixation rates of these mutations, as well as the probabilities of beneficial and deleterious alleles at fixed genomic sites. We find that interference interactions generate a regime of emergent neutrality: all genomic sites with selection coefficients smaller in magnitude than a characteristic threshold have nearly random fixed alleles, and both beneficial and deleterious mutations at these sites have nearly neutral fixation rates. We show that this dynamics limits not only the speed of adaptation, but also a population's degree of adaptation in its current environment. We apply the model to different scenarios: stationary adaptation in a time-dependent environment, and approach to equilibrium in a fixed environment. In both cases, the analytical predictions are in good agreement with numerical simulations. Our results suggest that interference can severely compromise biological functions in an adapting population, which sets viability limits on adaptive evolution under linkage.